
版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
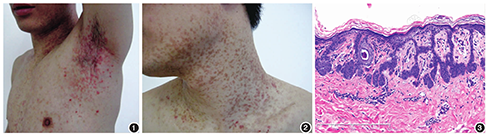
患者男,34岁,因面颈、躯干、上肢红丘疹及褐色斑6年就诊。患者6年前无明显诱因于颈部、双腋下、腹股沟陆续出现红色丘疹,炎症消退后呈现棕褐色斑片或斑丘疹,反复发作,之后累及前额、胸腹及后背,部分融合成网状,对称分布,有时瘙痒。体检:各系统检查未见明显异常。皮肤科检查:患者前额、颈部、躯干见成片棕褐色斑疹、斑丘疹,夹杂散在红色丘疹、斑丘疹,皮疹扁平,表面无鳞屑,孤立损害似脂溢性角化样;前额及颈部皮疹密集分布,融合呈网状;双腋下及腹股沟可见绿豆大小红色丘疹,呈密集网状分布,腰腹部皮肤可见弥漫性点状色素减退斑。见图1,图2。临床拟诊:疣状表皮发育不良、家族性慢性良性天疱疮、Darier病。分别取腹部棕褐色斑丘疹和颈部红色丘疹活检,组织病理类似:角化过度,棘层轻度增生,表皮突下延细长,呈芽蕾状或鹿角状,相互交织,见个别角囊肿,真皮浅层血管周围见少量淋巴细胞浸润,伴少数噬色素细胞(图3)。诊断:屈侧网状色素异常症。该例患者明确诊断后未予治疗,随访1年皮损未消退。
屈侧网状色素异常症又称Dowling-Degos病及黑点病,为一迟发性遗传性皮肤病,呈常染色体显性遗传,亦可散发。大部分患者发病在11岁以后,皱褶部位点状及网状色素沉着为该病主要特征,多始于腋下、腹股沟,之后发展至其他皱褶部位如乳房下、颈部,亦可累及手腕、前臂、大腿内侧、面部、头皮、外阴及阴囊等少见部位,进行性、对称性发病,颜色从棕色至黑色不等,可伴瘙痒。临床上需与Galli-Galli病、北村网状肢端色素异常症、遗传性泛发性色素异常症、遗传性对称性色素异常症、Darier病、Harber综合征、色素性痒疹以及融合性网状乳头瘤病鉴别。

























