Copyright © 2020 by Hospital for Skin Diseases (Institute of Dermatology), Chinese Academy of Medical Sciences and Chinese Medical Association
Please do not use or copy the layout and design of the journals without permission.
All articles published represent the opinions of the authors, and do not reflect the official policy of the Chinese Medical Association or the Editorial Board, unless this is clearly specified.
Fibrotic diseases comprise a group of disorders with a spectrum of clinical severity, including multisystem diseases, such as systemic sclerosis. In some conditions, the fibrosis is limited to specific organs, such as pulmonary, liver, kidney, and bladder fibrosis, as well as in skin in keloids.1,2,3,4,5,6 Although the pathomechanisms of these disorders may vary, the common unifying pathological feature is elevated expression of genes encoding extracellular matrix (ECM) proteins. The resulting fibrosis then perturbs the normal architecture of the affected organs and leads to their functional failure.7,8,9 Indeed, persistent production of ECM macromolecules by activated mesenchymal cells, often with characteristics of myofibroblasts, differentiates controlled repair taking place during normal wound healing from tissue fibrosis, the hallmark of fibrotic diseases.10,11,12,13 The chronic nature of the fibrotic processes and the large number of individuals suffering from their devastating effects emphasize the importance of these diseases as one of the most serious health problems globally. It has been estimated that as much as 45% of the mortality in the developed countries is caused by some form of fibroproliferative diseases,9 and the mortality in underdeveloped or developing countries caused by these diseases may be even higher. Emphasizing the importance of fibrotic diseases, there is currently no effective and specific treatment for this group of disorders.
In most cases, fibrosis is initiated as a reactive process, and a number of different factors can modulate the pathways leading to tissue fibrosis. Such factors, often propagated by the inflammatory tissue reaction, include the local expansion of resident fibroblast subpopulations, immune and microenvironmental modulation of the synthetic functions of fibroblasts, as well as altered regulation of various metabolic reactions governing the biosynthesis and turnover of the connective tissue components, primarily collagen. Thus, the net accumulation of collagen in tissue fibrosis is the result of an imbalance between the factors that either lead to enhanced production and deposition or impaired degradation of collagen.
Contributing to the metabolic modulation are cytokines and growth factors, a group of diverse proteins derived from blood cells, such as from platelets and leukocytes, or released locally by mesenchymal and epithelial cells. In addition to the complexity of the biosynthetic pathways and the multiple cell types recognized in fibrotic diseases, the degradation of ECM components, particularly collagen, has been well-established to involve a host of matrix proteases.
Keloid disorder, a recently coined term, refers to a group of fibroproliferative disorders, the keloids being the prototype, but also includes hypertrophic scars, keloidalis nuchae, and acne keloidalis.14 Keloids manifest clinically as solid papules, nodules, or plaques with the predilection sites on the face, chest, and upper back (Fig. 1). The development of keloids is often associated with trauma which, however, may be very slight or even unnoticeable. Keloids in the affected individuals cause considerable morbidity owing to their propensity to infections, erosions, and associated pruritus. Keloids can also be of considerable cosmetic concern with devastating psychosocial impact (Fig. 1).
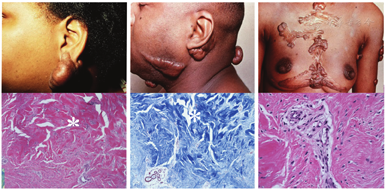
Keloids are particularly common in individuals with skin of color, and the estimated prevalence in populations of African ancestry is globally 4%-16%, that is, up to 15-fold higher than in Caucasians.15,16,17 Furthermore, individuals of Asian ancestry have a significantly elevated risk for keloids. These observations, together with familial clustering of keloids, suggest a strong genetic contribution to keloid formation.18,19 Genome-wide association studies have identified susceptibility loci for keloids in Japanese as well as in Chinese Han populations and suggested NEDD4 as one of the candidate molecules involved in keloid formation.20,21,22 This molecule has been suggested to enhance the proliferation and invasiveness of fibroblasts and activate the TGF-β/catenin transcriptional activity. Recent studies have also suggested that altered expression of microRNAs might partially contribute to etiology of keloids by affecting several signaling pathways relevant to scar formation and wound healing.23,24 Nevertheless, no candidate genes underlying keloid formation have been consistently identified, and how these genetic findings relate to pathogenesis of keloids remains currently unclear.
Histopathology of the keloid lesions demonstrates accumulation of ECM, primarily collagen fibers, which show compact packing (Fig. 1). Embedded into the matrix are variable numbers of fibroblastic cells, often with expression of marker proteins characteristic of myofibroblasts.25,26 Some progress has been made in understanding the composition and pathobiology of keloids. The primary genetic collagen types in keloids are type I and type III.27,28 In addition, type VI collagen has been shown to be upregulated at the early stages of keloid development, suggesting that type VI collagen may serve as an early biomarker of the fibrotic process.29 Nevertheless, candidate gene approaches have excluded TGF-β1-3, TGF-β receptors I, II, and III, as well as collagen proα2(I) as candidate genes harboring pathogenic sequence variants in keloid patients.30
Over the past two decades, significant progress has been made toward understanding the composition of keloids, characterized by excessive accumulation of collagen; however, the pathomechanistic pathways leading to ECM accumulation are less well defined. Previous observations have suggested that the activated myofibroblast is the key cell responsible for excessive ECM production, either directly or through paracrine action. However, the origin of activated myofibroblasts, the factors triggering excess matrix production by these cells, and the nature of downstream pathways resulting in skin fibrosis in keloids have remained less well characterized.
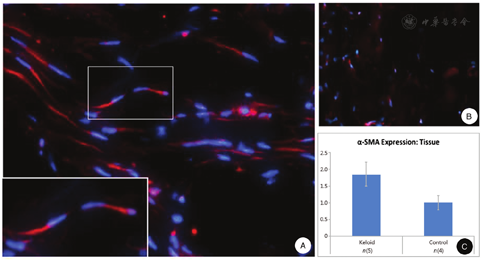
Recent studies focusing on other fibrotic diseases, particularly scleroderma, have provided novel, exciting insight into the pathomechanisms of fibrotic diseases in a manner that could be applicable also to keloids.31,32 Such information has suggested that activated myofibroblasts are derived predominantly from perivascular progenitor stem cells through a complex molecular process, and specifically, a subpopulation of perivascular cells with Gli1 as a specific marker are transitioned to cells characterized by the expression of myofibroblastic markers, α-smooth muscle actin (α-SMA), vimentin, and type I collagen. They, however, retain Gli1 expression as a marker of their perivascular origin. These Gli1+ myofibroblasts synthesize cellular fibronectin (cFN)-EDA, a fibronectin (FN) splice variant that signals through toll-like receptor 4 (TLR4) leading to excess production of ECM components and resulting in the development of tissue fibrosis.31,32,33,34
Early morphologic studies, based on light microscopy, immunohistochemistry, and electron microscopy, demonstrated considerable heterogeneity in cell populations within keloid lesions.35,36,37 Particular attention has been paid to cells with phenotypic features of myofibroblasts defined primarily by expression of α-SMA and vimentin (Fig. 2). These myofibroblasts depict prominent rough endoplasmic reticulum and Golgi apparatus, and presence of intracellular collagen within tubular membranes has been noted, suggesting activated biosynthetic and secretory capacity of these cells. In addition to myofibroblasts, the presence of myofibroblast-like subpopulations, with lesser degree of differentiation, have been described in keloids, attesting to the heterogeneity of the phenotypic features of myofibroblasts indicating cell plasticity of fibroblasts.25,38 In addition to myofibroblasts, Mast cells have been identified in keloids, often in close association with myofibroblasts with frequent direct cell-cell contacts.37 Keloids also contain a vascular component with endothelial cells being identified by Factor VIII-related immunoreactivity,39 and keloids have been shown to express vascular endothelial growth factor at high levels, suggested to be derived primarily from epidermis.40 Thus, there is considerable heterogeneity of the cell populations within the keloid lesions.
FNs, high-molecular-weight glycoproteins, are present in extracellular connective tissue matrices and extracellular fluids, including blood plasma. The human FN gene consists of 45 exons, and the primary RNA transcripts are alternatively spliced to form up to at least 20 different mRNA variants.41 The FNs interact with other matrix proteins, such as collagens, glycosaminoglycans, and fibrin, as well as cell surface receptors, including integrins α9β1, α5β1, α4β1, and αVβ3, as well as TLR4.42,43 The precise role of individual splice variants of FN in ECM biology and pathology remains largely unknown. However, one of the alternatively spliced exons encodes the extra domain (EDA) that is developmentally regulated and is found exclusively in cFN, which is produced by a variety of cells, including fibroblasts and epithelial cells, and is deposited as fibril matrices in the ECM.44
The in vivo role of the cFN-EDA variant has been previously studied by constructing transgenic mice either constitutively expressing (FN-EDA+/+) or excluding (FN-EDA-/-) the EDA segment of the FN polypeptide.45 Neither embryonic lethality nor postnatal malformation was observed in the case of homozygous mutant FN-EDA-/- mice, suggesting that EDA is not required for normal development. However, significant abnormalities were observed in adult FN-EDA-/- animals. Specifically, the EDA peptide segment was shown to be essential for normal wound healing, particularly with respect to re-epithelialization. Cellular responses to FN-EDA are carried out through binding to cell surface receptors, including TLR4 as well as integrins α4β1, α4β7, and α9β1.42,43
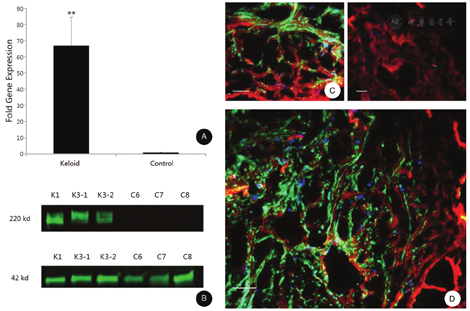
Compelling and exciting recent evidence has accumulated implicating a crucial role of cFN-EDA not only in normal wound healing but also in fibroproliferative disorders, particularly scleroderma.32 Specifically, Bhattacharyya et al.32 demonstrated increased cFN-EDA in the skin and circulation of patients with scleroderma. Additionally, exogenous cFN-EDA stimulated collagen production and myofibroblast differentiation in vitro mediated by TLR4. These experiments also demonstrated, by sequential immunoprecipitation, that cFN-EDA is an endogenous TLR4 ligand in scleroderma skin fibroblasts. The results revealed direct interactions of cellular TLR4 with cFN-EDA in unstimulated fibroblasts, and this was markedly increased in TGF-β treated cells. The importance of these studies is emphasized by the striking phenotypic differences between scleroderma and keloids, yet it is possible that similar cFN-EDA/TLR4 interaction-dependent pathways are the unifying feature in these two fibrotic diseases. These observations suggested, therefore, a role for cFN-EDA in the development of keloids, an abnormal fibroproliferative response elicited by trauma to the skin of genetically susceptible individuals. We have recently demonstrated a dramatic, up to 70-fold, increase in cFN-EDA expression in keloids (Fig. 3). The mechanisms of generation of activated myofibroblasts, and the specific molecular interactions between cFN-EDA and its endogenous receptors, particularly TLR4, represent a potential target for treatment development.
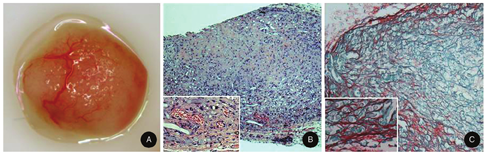
There are no spontaneous animal models for keloids. Consequently, we have focused on development of a graft model that consists of keloid cells embedded into a poly (lactic-co-glycolic acid) (PLGA) scaffold (Fig. 4). These implants have demonstrated vascularization, long-term survival, and evidence of cell viability at least up to eight weeks after grafting subcutaneously into Rag1-/- mice (Fig. 4). These grafts depict accumulation of collagen, as demonstrated by histopathological stains (H&E and Sirius red; Fig. 4B and Fig. 4C).
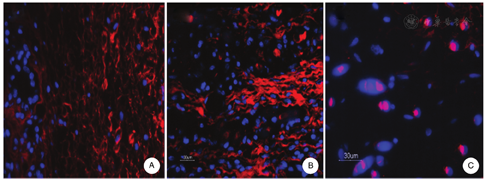
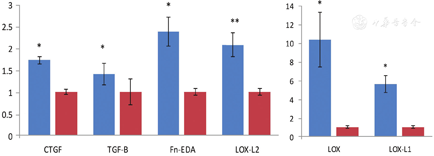
Collagen that accumulated in the graft was shown by immunofluorescence labeling to consist of type I and type III collagens (Fig. 5A and Fig. 5B), and importantly, keloid scaffolds stained with an antibody to Gli-1 showed intranuclear staining (Fig. 5C). The grafts also expressed higher levels of pro-fibrotic proteins, particularly connective tissue growth factor, TGF-β and FN-EDA, as well as lysyl oxidase and lysyl oxidase-like (LOX, LOX-L1, and LOXL-2) proteins than grafts seeded with fibroblasts from control skin (Fig. 6). We have used this model system to study the effects of an antibody directed against the telopeptide region of human type I procollagen in a manner that it interferes with the packing and cross-linking of newly synthesized collagen.46,47
Treatment attempts to remove and/or counteract keloids are often suboptimal.48,49,50,51 For example, surgical removal of these lesions is frequently followed by reoccurrence, which either compromises the results or makes the outcome even worse, although radiation therapy may be helpful in preventing the regrowth of the lesions. Thus, radiation therapy can play an important adjuvant role in the treatment of keloids.52,53,54 Intralesional steroid injections can lead to softening and partial resolution of keloids, however, with incomplete end result. The mechanisms of action of intralesional steroids have been suggested to involve downregulation of collagen gene expression and upregulation of matrix metalloproteinases, synergistically resulting in dissolution of the collagen meshwork. In addition, a number of cytostatic or cytotoxic agents, CO2 laser treatment, freezing, topical silicone sheaths, pressure therapy, and so on have been tested with variable results.51,55
Keloids are common fibroproliferative tumors with considerable morbidity in the affected population, yet with suboptimal treatment options. These lesions are characterized by accumulation of ECM of connective tissue, particularly collagen, primarily elicited by mesenchymal cells with myofibroblastic differentiation. The collagen biosynthetic machinery is activated by a number of cytokines, particularly TGF-β. In addition, a number of ECM proteins, such as the EDA splicing variant of cFN, contribute to the fibrogenesis resulting in formation of the keloid lesions. Understanding of the mechanistic details of the collagen biosynthesis may provide critical information for development of novel treatment modalities for fibrotic diseases, as exemplified by keloids.
This study was supported by the institutional funds of the Sidney Kimmel Medical College.
Carol Kelly assisted in manuscript preparation.
The authors reported no conflicts of interest.

























