
分析总结我院46,XX性发育异常患儿性腺探查结果并分析。
回顾性分析2006年1月至2015年12月期间就诊我科染色体核型为46,XX的性发育异常患儿98例,其中24例因内分泌科明确诊断CAH无需行性腺探查。另74例46,XX DSD为明确诊断行手术探查性腺,并取病理活检。根据术中探查所见内生殖器分布情况以及术后患儿的病理结果汇报归纳总结74例46,XX患儿性腺探查情况并分析。
除CAH外余74例46,XX DSD患儿中社会性别为:男48例,女26例。为明确诊断均行性腺探查术。其中单纯腹腔镜手术49例,腹腔镜联合开放手术12例,开放手术13例。性腺探查病理结果如下:双侧性腺均为卵睾25例(33.78%);一侧性腺为卵巢,一侧为睾丸13例(17.57%);一侧卵睾,一侧睾丸12例(16.22%);一侧卵睾,一侧卵巢18例(24.32%);双侧均为原始性腺2例(2.7%);一侧原始性腺,一侧卵巢2例(2.7%);一侧原始性腺,一侧睾丸2例(2.7%)。31例含有卵巢性腺患儿中14例卵巢分布在左侧,占45.16%。25例含有睾丸性腺的患儿中11例睾丸位于右侧,占44%。
性腺探查及病理活检是46,XX DSD中除CAH外明确性发育障碍患儿的诊断及治疗方案重要的评估手段,对早期明确诊断、合理的选择性别及手术重建治疗具有重要的意义。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
性发育异常或先天性性别异常(disorders of sex development,DSD)是指由于染色体、性腺及性解剖结构异常等造成的一类先天性疾病。根据染色体不同分为性染色体DSD、46,XY DSD和46,XX DSD。其中46,XX DSD是性别发育障碍中最常见的类别,DSD的不同病因可有相同或相似的临床表型,同一病因发生时间不同或影响程度不同所致的临床表型差异亦甚大,加之目前医疗诊断技术有限、家长认知误区,故临床明确诊断并合理治疗仍是目前儿科临床的棘手难题,尤其是儿童阶段的早期诊断,临床上容易出现误诊或漏诊,这将直接影响患儿的治疗策略及其预后。性腺探查病理活检是明确其内生殖器中性腺成分的金标准,是后期治疗的基础。本文将回顾性分析74例46,XX DSD患儿性腺探查的结果。
回顾性分析2006年1月至2015年12月期间就诊我科染色体核型为46,XX的性发育异常患儿共98例,社会性别男50例;社会性别女48例(其中24例CAH患儿中社会性别女22例,社会性别男2例)。就诊年龄1~12岁,平均年龄6.5岁。就诊时家长主诉:外生殖器异常85例;双侧隐睾并阴茎发育差4例;单侧隐睾合并小阴茎3例;因疝气手术发现异常性腺2例(1例为嵌顿疝急诊手术发现异常性腺,另1例为女孩择期手术时发现睾丸),重度尿道下裂4例。就诊患儿中社会性别为男性者均存在不同程度尿道下裂。其中24例经内分泌科已明确诊断CAH,无需性腺活检,因Prader分级为Ⅲ级及以上,故予以行阴蒂短缩外阴成形术。另74例患儿外生殖器表型模糊,诊断不清,予以行手术探查性腺。其中单纯腹腔镜手术49例,腹腔镜联合开放手术12例,开放手术13例。探查术中双侧性腺全部取病理送检以明确性腺成分。
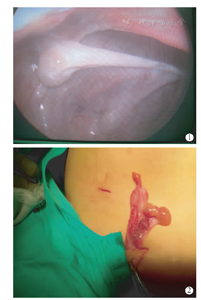
74例均予以行性腺探查术。术后病理结果如下:双侧性腺均为卵睾25例(33.78%)(图1为腹腔镜下所见;图2为腹腔镜联合开放手术所见);一侧性腺为卵巢,一侧为睾丸13例(17.57%);一侧卵睾,一侧睾丸12例(16.22%);一侧卵睾,一侧卵巢18例(24.32%);双侧均为原始性腺2例(2.7%);一侧原始性腺,一侧卵巢2例(2.7%);一侧原始性腺,一侧睾丸2例(2.7%)。31例含有卵巢性腺患儿中14例卵巢分布在左侧,占45.16%。25例含有睾丸性腺的患儿中11例睾丸位于右侧,占44%。
另24例患儿因门诊已经内分泌科明确诊断CAH,无需性腺活检,均按女孩抚养。但因Prader分级为Ⅲ级及以上,故予以行阴蒂短缩外阴成形术。术后患儿恢复好,外观基本满意,无排尿困难、尿失禁等不适。
胚胎时期的性发育主要包括性别决定、性腺、内生殖管道和外生殖器的分化和发育,是一个复杂而又连续的有序过程。根据Jost的理论[1],主要包括以下三步:首先在受精时确定染色体,这决定性腺是向睾丸分化还是卵巢分化,然后在相应的性腺的内分泌作用下,引导内部生殖管道的分化和外生殖器的形成。因此性别表型的发展是遗传信号和激素信号相互作用的结果。已知正常性腺分化发育主要依赖于三方面功能正常[2]:①决定个体的遗传性别性染色体(XY和XX);②宫内调节性器官分化发育的相关因子;③下丘脑-垂体-性腺(H-P-G)轴功能。其中涉及性别导向决定的调控及其性腺分化发育的一系列级联程序,是由多种相关基因参与的复杂过程。任何环节发生异常均可导致性腺及性器官异常发育分化,导致临床出现性发育异常或先天性性别异常(disorders of sex development,DSD)。2006年,美国儿科内分泌协会和欧洲儿科内分泌协会的许多专家学者提出,将染色体性别、性腺性别或表型性别发育异常者统称为性发育障碍[3],摒弃原有界定混淆又加剧患儿心理负担的诸如两性畸形、间性畸形、性反转等命名方式。按照染色体核型大致分为性染色体DSD、46,XY DSD和46,XX DSD三大类。46,XX DSD是性别发育障碍中最常见的类别,具有46,XX正常女性染色体核型、卵巢和苗勒管衍生物。在外生殖器发育过程中,由于受到内源性或外源性雄激素的影响,可有不同程度男性化表现,引起外生殖器性别模糊[4]。关于46,XX DSD可能发病的机制有多种学说,目前占主导的主要三种:①SRY基因易位(如46,XX卵睾型DSD)[5];②胎儿期促进性腺发育和分化的相关因子异常(如Wnt-4基因突变);③雄激素过量(如CAH中21.羟化酶缺乏症)。雄激素过剩是最常见的46,XX DSD,以往被称为先天性肾上腺皮质增生(CAH)[6]。本病是一常染色体隐性遗传疾病,由于皮质醇合成过程中某一种必需的酶缺陷导致皮质醇的合成不足,反馈促使促肾上腺皮质激素释放激素和促肾上腺皮质激素代偿性增加,引起肾上腺皮质的增生,增生的皮质网状带分泌过高的雄激素而出现外生殖器发育异常(男性化)。常见的酶缺陷依次为:21-羟化酶,11β-羟化酶,17α-羟化酶等。其中,21-羟化酶缺陷最常见,约占95%。本组病例中98例46,XX DSD中CAH占24例,占24.49%。此比例明显低于国内外文献报道的比例,这是因为我院CAH的诊断在内分泌科进行,仅Prader分级为Ⅲ级及以上需外科手术干预患儿就诊我科,准确地讲这一比例应为Prader分级为Ⅲ级及以上CAH所占比例。事实上,CAH应为46,XX DSD最常见的一类。
正常的SRY基因易位到X染色体或常染色体上是导致46,XX DSD另一个主要原因。46,XX卵睾性DSD是其中一种比较特殊的疾病,随着对人类基因研究的进展,国内外近年报道逐渐增加[7]。其外生殖器可为男性阴茎(或有尿道下裂,或有小阴茎,或有勃起屈曲)和睾丸(或细小,或隐睾,或仅有一侧,或为卵睾),女性男性化表现,外生殖器模糊。其性腺类型为卵睾体,多位于正常卵巢位置且多在右侧;如卵睾体内睾丸组织多于卵巢组织易发生卵睾体下降至阴唇、阴囊或腹股沟内;多数一侧为卵睾体,另一侧为卵巢(多在左侧);卵巢和(或)卵睾体内的卵巢组织多正常发育且可见黄体,而睾丸或卵睾体内的睾丸组织常不同程度发育不良。本文74例患儿性腺探查结果中我们发现卵睾型DSD占的比例最高。
既往国外文献报道[8]异常的性腺畸形包括3种形式:①双侧型即两侧均为卵睾,约占30%;②单侧型即一侧为卵睾,另一侧为睾丸或卵巢,约占50%;③片侧型即一侧为睾丸,另一侧为卵巢,约占20%。本组研究中片侧型占17.57%;单侧型:一侧为卵睾;一侧为卵巢占24.32%、一侧为睾丸占16.22%;双侧型占33.78%。与既往大组病例的统计基本相符。崔佳等[9]亦报道双侧型占21.4%,单侧型与片侧型各占42.9%、35.7%。Krob等[10]也曾对283例DSD患儿性腺分析发现卵睾所占比例最多达44%,其次是卵巢占21%,睾丸占12.5%。并且我们发现患儿性腺分布依然遵循了卵巢多在左侧,睾丸或卵睾多在右侧的规律[11]。本文74例患儿中含有卵巢性腺的31例中14例卵巢分布在左侧,占45.16%。25例含有睾丸性腺的患儿中11例睾丸位于右侧,占44%。性腺的不对称发育是由遗传因素和环境因素共同决定的,但其遗传机制目前尚不清楚[12]。
对患儿的性腺和外生殖器进行完善的临床评估是非常重要诊断环节,这对性别选择、手术干预、长期治疗策略制定等都具有重要意义。临床评估主要包括:①表型:观察患儿有无性征的表型男性化或女性化的异常表现,有无相貌、骨骼等的伴发畸形及其智力水平等。本文98例患儿中,外生殖器表现各异,男性患儿均有不同程度的尿道下裂。女性多表现阴蒂肥大;②性腺触诊:尤其表型女性化伴腹股沟斜疝者,需要触诊确定有无睾丸及其位置、大小、质地。检查患儿的外生殖器状况时应按照Prader分类标准[13](从正常女性到正常男性之间可分为I~V型)进行分类,以评估外生殖器的解剖状况是接近男性还是女性。如此评估能够比较直观地了解患儿的外生殖器状况,并能在随访中客观且标准化地记录患儿外生殖器的演变情况,对于之后的性别选择、手术方式的选择以及激素替代治疗的选择都有重要意义。
DSD患儿的外科干预主要应用在两方面:性腺探查及重建手术。性腺探查及病理活检是明确性发育障碍患儿内生殖器情况的重要方法。依据术前的诊断和评估,术中应仔细寻找异常性腺的可能藏身之处。如果术前考虑睾丸,则重点寻找腹股沟管内口处,尝试牵拉通入其内的韧带,往往能够成功。如果考虑为条索状性腺,则仔细寻找纤细的输卵管和发育不全的始基子宫,与输卵管平行相伴的鱼白色细条索很可能就是异常的性腺。腹腔镜手术是目前最常用的探查方法[14],对于双侧腹股沟区可触及的性腺可采用开放手术。
DSD患儿的外科重建手术主要是基于以下因素进行:①改善外生殖器外观;②获得无阻碍的具有性别特异的排尿方式(如男性可以站立排尿);③完成阴道阴茎性交。虽然DSD外科手术已经探索了十多年,但目前如何进行性别选择,如何选择手术时机仍然存在有很大的争议[15]。支持婴儿期即进行手术的学者认为,婴幼儿期操作比较简单,减轻患儿生殖器模糊及家属生活上的耻辱感。支持晚些进行重建手术的学者认为:①手术更适合患儿决定而不是家属;②目前国内外尚没有数据可表明早期生殖器手术和性心理取向之间的联系[16]。文献报道[17]患儿到青春期或者成人后对手术效果不满意比例可高达47.1%,对性功能不满意可达37.5%,性焦虑者高达47.4%[18]。在一项研究中33例生后生殖器模棱两可的患儿中41%的早期行手术,但待患儿成年后并不满意其外生殖器或者性别的选择,89%需要重新手术,39%重新行阴蒂再生术[19];③生殖器重建手术方案的选择应该以保护原则为先,以防最初的性别判断是错误的和有性别改变的需求;④有的DSD患儿并没有行生殖器成形术的需求,无需早期手术[20]。本组74例经腹腔镜性腺探查明确诊断后经家长慎重考虑坚决要求按男性抚养,切除女性性腺,成型尿道41例(55.4%);按女性抚养,切除男性性腺,阴蒂成型19例(25.67%);14例(18.92%)患儿目前尚未做出选择,未做后续治疗。但因目前尚未进行临床随访,需在今后的研究中进一步随访。
无

























