
探讨先天性血管瘤(CH)患儿的临床及病理学特征、诊断、鉴别及治疗。
对32例行手术切除CH患儿的临床资料、病理结果及治疗进行总结。
患儿男20例,女12例,男女比例为1.6∶1,平均就诊年龄3岁7个月。均为单发肿块,头颈部8例,躯干12例,上肢5例,下肢7例。1例为产前B型超声发现,余31例为患儿出生时发现。25例肿块有逐渐增大表现;3例肿块较出生时有缩小;4例肿块大小无明显变化。1例肿块触诊皮温较高,1例肿物中央可及一窦道,余30例均无局部疼痛、红肿破溃等情况。所有病例术前血常规及凝血谱检查均未见异常。1例患儿曾接受硬化剂注射术,术后肿块无明显缩小。所有病例均行手术完整切除。1例患儿术中出血过多,术后接受输血治疗。结合临床表现后诊断:快速消退型先天性血管瘤(RICH )3例,余29例为不消退型先天性血管瘤(NICH)。NICH:镜下见瘤组织呈小叶状结构,小叶内微血管内皮细胞核呈"靴钉样"向管腔内突出。小叶中央见星形管腔,间质为致密纤维组织,可见淋巴管结构及明显的小动、静脉。RICH:镜下见与NICH相似的小叶状结构,小叶内多量血管闭锁,余血管腔变窄,内皮细胞较扁平,小叶间纤维脂肪组织增多,可见灶性出血坏死、钙化及含铁血黄素沉着。免疫组织化学染色:所有病例均不表达Glut1,均表达CD31、CD34、WT1,D2-40部分表达。随访25例(随访时间3个月至2年1个月),可见残留轻微线样瘢痕,均未见复发。
CH属良性脉管肿瘤,预后较好。诊断RICH/NICH时需结合临床表现和病理形态。病理上需与脉管畸形、婴儿性血管瘤、卡波西形血管内皮瘤及化脓性肉芽肿相鉴别。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
先天性血管瘤(congenital hemangioma,CH)是指在宫腔内基本发育成熟,出生后不再进展的一类血管肿瘤,Glut1染色阴性,是婴幼儿较少见的良性脉管肿瘤,分快速消退型先天性血管瘤(rapidly involuting hemangioma,RICH)和不消退型先天性血管瘤(noninvoluting hemangioma,NICH)及部分消退型先天性血管瘤(partially involuting hemangioma,PICH)3种亚型,三者除临床病程不一致外,在发病部位、性别比、影像表现及组织形态方面均相似。目前国内关于CH的报道少见,现本文对32例CH的临床特征及病理学特点进行总结,并结合相关文献进行分析,以进一步加深对该肿瘤的认识,有助于诊断和治疗。
收集浙江大学医学院附属儿童医院于2014年1月至2016年1月间手术切除的CH标本32例。其中RICH 3例,NICH29例。临床资料来自于电子病例,影像学资料来自PACS-WEB系统。
32例中,男20例,女12例,男女比例为1.6:1,就诊年龄最小者3个月,最大者10岁,平均年龄3岁7个月。发病部位:头颈部8例,躯干12例,上肢5例,下肢7例。均为皮肤单发肿块,1例为产前B型超声发现,余31例为患儿出生时发现。22例呈青紫色或紫红色隆起型肿块,周边可见扩张营养血管;4例为红色斑块样,触诊时皮下可及包块;6例突起肿块表面肤色正常。25例患儿发病后肿块有逐渐增大表现;3例患儿肿块较出生时有缩小;4例肿块大小无变化。1例肿块触诊皮温较高,1例肿物中央可及一窦道,余30例均无局部疼痛、红肿破溃等情况。所有病例术前血常规及凝血谱检查均未见异常。21例行B型超声检查,显示低回声包块,内部有不均匀回声,见较丰富的血流信号。3例患儿进行MRI检查,见T1WI与肌肉等信号,T2WI呈高信号,可见血管流空信号。1例患儿在外院行CT检查,显示皮下巨大软组织密度肿块影,伴多囊变。1例患儿曾在外院接受硬化剂注射术,术后肿块无明显缩小。所有病例均行手术完整切除。1例瘤体较大患儿术中出血较多,术后复查血常规,见Hb仅80g/L,给予输红细胞悬液80ml。余患儿未见出血过多、术后贫血及血小板减低等情况。
所有标本经4%中性缓冲甲醛溶液固定、石蜡包埋、4 μm厚切片、HE染色。请主任医师复核本组所有病例的HE切片及免疫组化切片。
采用EnVision法,所用抗体及克隆号:CD31(JC/70A)、CD34(QBEnd/10)、Glut1(多克隆)、D2-40(D2-40),WT1(6F-H2),抗体购自Dako或福州迈新生物技术开发有限公司。按产品说明书的操作步骤进行染色。所有标本均行CD31、CD34、D2-40、Glut1及WT1染色,实验时同时设置阴性及阳性对照。
肿块大小不等,最小者1.5 cm×1.0 cm×1.0 cm,最大者达7.5 cm×4.0 cm×3.5 cm,肿瘤表面暗红色或灰红色,部分病例皮肤粗糙。切面:灰红灰白色,质中。
29例NICH:见瘤组织呈小叶状结构,小叶大小不等、形状不一,由大量微血管组成,衬以内皮细胞,细胞核呈"靴钉样"向管腔内突出,小叶中央见星形管腔,间质为致密纤维组织,可见明显的小动脉及小静脉,散在淋巴管结构。其中6例见管腔内充红细胞,但未见血栓形成;5例见管腔内充淋巴液;2例见小灶钙化。3例RICH:见瘤组织呈小叶状结构,小叶间纤维脂肪组织增多,大部分血管闭锁,余血管腔变窄,内皮细胞较扁平,可见小灶性出血坏死、钙化及含铁血黄素沉着。
3例RICH和29例NICH均表达CD31、CD34、WT1,D2-40散在阳性,Glut1均阴性。
以电话方式进行随访,截止至2016年1月,随访时间3个月至2年1个月。32例随访到25例,可见残留轻微线样瘢痕,但均未见复发。
CH的概念最早由Boon于1996年提出[1],用于描述一类在母体子宫内增殖,出生时基本发育完全,婴儿早期开始消退的血管瘤。随后North于2001年提出先天性非进展性血管瘤的概念[2],指出该瘤的生长曲线与普通的婴儿性血管瘤的增殖曲线不同,且组织形态及免疫组化特征也相异,为一类葡萄糖转运蛋白1(Glucose transport-1 protein,Glut1)阴性血管瘤。2006年国际脉管异常研究组织(International Society Of The Study Of Vascular Anomalies,ISSVA)按其不同的生物学特性,分出快速消退型先天性血管瘤(RICH)和不消退型先天性血管瘤(NICH)两个亚型[3]。RICH一般在出生6~14个月内完全消退[1],偶有病例亦可宫内消退[4,5]。而NICH持续存在,不会自行消退,大小不变或者随患儿身体长大而有轻微地增大。近年来又新分出部分消退型先天性血管瘤(PICH)[6]。PICH在出生后一年内肿块类似RICH开始消退,然后进入缓慢的消退,该过程可持续12~30个月,平均18个月,最终病灶残留,呈NICH样肿块[7]。
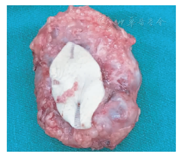
先天性血管瘤的确切发病率仍不清楚,但明显低于婴儿性血管瘤的发病率。本组收集的32例,约占同期所有血管瘤病例的10.3%,占同期脉管异常病例的6.7%,而同期IH所占比例分别为67.5%和44.3%。男女发病率无明显差异。大体一般表现为孤立性的红色、青紫色斑片样皮下浸润性生长或外生型肿块,直径几厘米至十几厘米不等(图1)。好发部位为头、颈及四肢近关节处,偶见肝脏[8]及颅内发病的报道[9,10]。
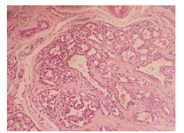
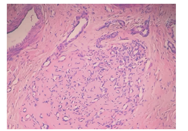
瘤组织常发生在真皮层和/或皮下组织,呈小叶状结构,小叶大小不等、形状不一,由大量微血管组成,微血管大小不一、形状不规则,腔内衬以内皮细胞。内皮细胞胞质常不明显,核突出,似"靴钉样"(图2)。小叶中央见星形管腔,管壁厚薄不一。间质为致密纤维组织,可见淋巴管及明显的小动静脉。核分裂象少见。可见肥大细胞数量增多[11]。而RICH与NICH相比,同样的小叶状结构内可见多量的血管闭锁、纤维化,余血管腔狭窄(图3),内皮细胞扁平,核不突出,间质见更多的纤维脂肪组织,血栓、含铁血黄素沉着、灶性钙化可见。
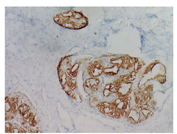
Glut1在IH的各个时期均有表达,而各型CH均不表达[12],是CH与IH鉴别的重要工具。最近研究显示WT1(Wilms Tumor 1)在内皮增生的脉管肿瘤内阳性表达,而脉管畸形中除动静脉畸形增生期表达外其余类型的脉管畸形不表达[13],是鉴别脉管肿瘤与绝大多数脉管畸形的有效工具。本组病例WT1均呈阳性表达,且表达部位在胞浆(图4),与文献报道相一致。D2-40是淋巴管内皮细胞特异性标志,由于CH瘤组织内散在淋巴管结构,因此可见血管小叶边缘阳性而小叶内阴性的形态特征,可作为诊断的参考指标。
CH的增生期发生在宫腔内,因此产前高分辨率超声即可发现,可见最早在胎龄12周超声诊断的报道[1]。但能否发现肿块与胎龄、胎儿姿势、肿块的位置及大小、检查者的技术水平密切相关,因此宫内的检出率并不高。Boon等[1]报道23例患儿行产前超声检查,仅检出3例。本组患儿仅1例为产前超声检出。而出生后的肿块结合其临床表现与影像学检查,即能诊断。部分影像检查不能诊断者,可以考虑病理活检。CH的影像特征与IH有部分重叠,但与IH相比,CH可见更多的血管、钙化及脂肪浸润,界限欠清。而其中NICH比RICH含有更多的血管分支,RICH钙化率高于NICH[14] 。
组织学上,CH需要与以下脉管异常相鉴别:①婴儿性血管瘤(IH):免疫标记Glut1阳性具有鉴别价值。IH多在出生3~6个月后快速生长,后进入缓慢的消退期,7岁时最终消退率达76%[15],镜下可见病变血管浸润周围神经及汗腺等组织;而CH为Glut1阴性血管瘤,镜下瘤组织不浸润至周围神经[2],其中RICH能快速消退;NICH不能消退,持续存在;PICH消退不全,最终类似NICH样肿块;②脉管畸形:病变一般出生时即有,肿块会随患儿的长大而呈比例性地增大。镜下见发育异常的脉管,无血管小叶的结构,内皮细胞无"靴钉样"表现,核分裂象少见。MRI常可见低信号的静脉石,无血管流空信号。Glut1染色阴性,WT1除动静脉畸形(增生期)阳性表达,其余类型的脉管畸形均不表达[13];③卡波西型血管内皮瘤:镜下可见结节,结节内为梭形肿瘤细胞及裂隙状血管腔,结节间为致密纤维结缔组织,其中部分KHE可见典型的肾小球样结构。肿块边界不清,常浸润至周围组织。是伴有卡-梅现象(kasabach-Merritt Phenomenon,KMP)的血管瘤的主要病理形态。而CH内无梭形肿瘤细胞,且伴KMP的病例罕见[1],也有学者认为CH偶有病例见短时凝血障碍及血小板减低,但其严重性不能与KMP相比[16]。本组病例均无KMP;④化脓性肉芽肿:为获得性、反应性毛细血管增生性病变,可以发生于任何年龄,常发生在易受外伤部位。镜下见毛细血管呈簇状或小叶状,可见大量新生毛细血管及炎细胞浸润,类似肉芽肿。
CH的临床处理方式与患儿年龄、肿块的部位、大小、深度、类型及是否有合并症相关,另外尚需考虑患儿及家属的心理负担,应区别处理。理论上,无并发症的RICH一般不需要治疗,连续随访观察即可。但该瘤的触诊感觉与卡波西型血管内皮瘤较相似,在行活检前,病理类型不明,临床恐有卡-梅综合征风险,因此,本组1例瘤体较大的婴儿直接选择手术完整切除。另2例RICH患儿肿块消退后残余皮肤影响美观,同样选择外科重建。3例RICH患儿手术年龄分别为11个月、9个月及1岁。但金云波等[17]发现RICH快速消退后肿块残留的皮肤皱缩及皮表颜色异常,会随观察时间的延长而改善,逐渐接近正常皮肤的质地和颜色。因此,临床上应慎重把握随访时间和手术时机。偶见RICH合并充血性心力衰竭[18]、皮表溃疡引起致命性出血[19] 、PHACE综合征[20]、皮肤脓疱[21]、短时内血小板减低[22,23]等并发症的报道,此类有合并症的患儿也需对症处理。NICH肿块持续存在,不消退,影响美观及患儿心理发育,临床上主要选择外科切除术。术中常规电凝止血,术后应用邦亭或巴曲亭等止血剂。仅1例患儿肿块较大,术中出血较多,术后轻度贫血,接受输血治疗。有学者提出硬化剂治疗后手术切除[24]。而本组1例患儿曾接受硬化剂注射,结果无效。若病灶较大,可能出血较多,可选择术前栓塞术[15]。尚可借助脉冲染料激光[16]及冷冻疗法治疗[25] ,但这两种方法治疗并发症多,如瘢痕、局部破溃出血、治疗不彻底、功能障碍等,临床应严格把握治疗适应指征[26]。本组25例患儿获得随访(随访时间3个月至2年1个月),均未见复发,或与随访时间较短相关,术后遗留较轻微的线样瘢痕,在可接受范围内。
无

























