
本综述介绍了NTDs(神经管缺陷)的疾病特点,总结了目前国际及国内范围对该病的病因研究情况,综合近五年的对该病的表观遗传发病机制研究的文章,分别从DNA甲基化、组蛋白修饰、印迹基因、小分子核糖核酸等表观遗传修饰在NTDs病因中的作用进行了详细的描述,并从NTDs发病的相关信号通路入手,总结了不同信号通路中基因组DNA甲基化与NTDs发生的关系。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
神经管缺陷(Neural tube defects, NTDs)是神经系统的先天性出生缺陷疾病,由胚胎发育早期神经管闭合失败引起,主要表现为无脑畸形、脑膨出、脊柱裂、脊髓脊膜膨出等,其发病机制尚未明确。世界范围内NTDs的平均发生率为1‰[1],我国是NTDs的高发地区,尤其是山西省,发病率高达13.87‰[2]。据中国卫生部发布的出生缺陷防治报告[3],我国每年新发NTDs高达1.8万,给患儿、家庭及社会带来了巨大的压力和负担。虽已证实孕早期补充叶酸可使NTDs发生率明显降低[4],但并不是所有的个体均对叶酸敏感;此外,神经管约在胚胎28 d完成闭合[5],此时大多数妇女还未意识到怀孕。因此,有部分妇女补充叶酸后仍育有NTDs子代。基因是人体遗传物质的载体,基因突变导致NTDs发生的研究已有很多,如:叶酸代谢相关基因(亚甲基四氢叶酸还原酶(MTHFR)[6]、亚甲基四氢叶酸脱氢酶[7]等)及其他一碳单位代谢通路中的关键酶基因(甜菜碱同型半胱氨酸甲基转移酶[8]、胱硫醚合酶[9]、甲硫氨酸合成酶还原酶[10]等)变异。在神经管汇聚延伸过程中平面细胞极性(PCP)信号通路发挥不可代替的作用,已有研究[11,12,13]证实PCP信号通路中的Vangl1、Fzd3、Fzd6等PCP核心基因突变可增加NTDs的发生风险,经典的Wnt信号通路也与NTDs的发生密切相关[14]。除了上述的营养、基因及其交互作用外,表观遗传调控也参与NTDs发生[15]。表观遗传学是受环境因素影响的、对基因表达有调节作用的一种调控机制,它包括但并不局限于DNA甲基化、组蛋白修饰、基因印迹、非编码小RNA分子调控。随着表观遗传修饰突飞猛进的发展,NTDs发生的表观遗传因素日益成为研究的焦点。
DNA甲基化是指不改变DNA的核苷酸序列而影响基因表达的一种表观遗传修饰,它主要发生在胞嘧啶的第5位碳原子上,在DNA甲基转移酶的介导下,使S-腺苷甲硫氨酸提供的甲基与胞嘧啶的碳原子共价结合。DNA甲基化可改变基因表达水平,从而使基因表型发生变异。DNA甲基化作为一项重要的表观遗传修饰,在维持染色体结构,X染色体失活,基因印迹和肿瘤发生中发挥重要功能[16]。DNA甲基化是可逆的且受环境因素影响,甲基供体不足时导致DNA甲基化模式发生变异。一碳单位代谢途径是甲基供体的主要来源,因此一碳单位代谢异常(如:叶酸、同型半胱氨酸、甜菜碱、维生素B2、维生素B6等供应不足或代谢异常),可导致DNA甲基化异常。
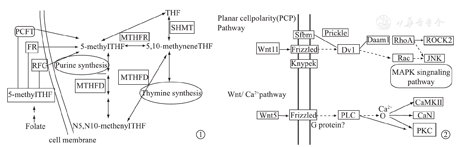
神经管发育始于胚胎第3周,历经神经板隆起,神经褶靠拢融合,神经孔闭合等形态发育,在此期间不仅涉及代谢通路对神经管发育的营养供给,如叶酸代谢提供DNA合成的原料(图1),还涉及其他相关通路(图2),如PCP信号通路在汇聚延伸运动中发挥重要作用,表现为神经板隆起形成神经褶并向中线靠拢融合发育成神经管,Wnt信号通路在神经胚形成,细胞极性建立等过程发挥作用。
目前,NTDs相关的DNA甲基化研究已引起广泛关注,其中,叶酸代谢相关的基因甲基化无疑成为关注的重点。MTHFR是叶酸代谢的关键酶基因,MTHFR基因甲基化研究结果尚不一致,有学者认为MTHFR多态性(C677T)与DNA甲基化异常相关[17],C677T突变导致的DNA甲基化异常是NTDs的病因之一,鉴于此观点尚不统一,Wang等[18]进行了Meta分析,结果显示MTHFR基因多态性与DNA甲基化改变之间尚无相关性。
叶酸在空肠上端被吸收后进入细胞主要通过3种类型的受体介导:还原型叶酸载体(RFC)、质子-耦合叶酸转运蛋白(PCFT)、叶酸受体(FR)。RFC是在小肠组织中表达的叶酸运输到细胞必不可少的叶酸载体。当叶酸在小肠中吸收时,主要通过RFC途径完成,已证实RFC基因80A>G多态性增加NTDs发生风险[19]。PCFT是一种高亲和力的质子-耦合叶酸运载体。FR是叶酸膜受体,通过介导内吞机制转运叶酸。叶酸转运基因表观研究[20]显示营养因素可能对RFC基因DNA甲基化产生影响,相对低水平同型半胱氨酸的个体,高水平同型半胱氨酸的个体的DNA甲基化水平较低;并且发现在NTDs胎盘中RFC、FR基因的差异性甲基化区(DMR)参与其表达调控,二者的高甲基化片段与mRNA低表达相关,此外,还发现RFC基因DNA甲基化对甲基供体缺乏比其他基因(PCFT、FR)更敏感。RFC基因80A>G多态性影响RFC基因的甲基化,证实了在NTDs中营养-遗传-表观遗传的交互作用。
Vangl基因和Fzd基因作为PCP信号通路的核心基因,在神经管汇聚延伸过程中至关重要。Vangl基因的甲基化研究[21]显示NTDs患儿脑组织Vangl基因的DNA甲基化水平较对照组高,但并无统计学意义,尚不能证实Vangl的DNA甲基化水平与NTDs发生有关。Fzd3的DNA甲基化研究[22]发现脊柱裂组的平均甲基化水平较对照组高,且差异有统计学意义(13.7%比10.91%,P=0.001),Fzd3基因包含R1、R2、R3三个亚区,其中分析了R2亚区的DNA甲基化水平与脊柱裂风险间的关系:高水平的DNA甲基化增加脊柱裂的发生风险(OR=11.8,95%CI: 2.03~13.32),在控制胎龄和性别后,这种相关性仍存在(AOR=10.5,95%CI:1.91~13.38),表明Fzd3基因R2亚区DNA甲基化与脊柱裂发生相关,进一步研究证实,DNA高甲基化参与Fzd3基因表达下调,支持了DNA甲基化在Fzd3基因表达调节中的作用,提示Fzd3启动子DNA甲基化增高,特别是R2区域的DNA甲基化增高引起的转录抑制可能是脊柱裂发生的危险因素。
Wnt信号通路在NTDs形成中起重要作用,不仅涉及胚胎背面-腹面轴的形成,而且在细胞极性的建立中发挥重要作用。Wnt信号通路中的两个基因(Siah16、Prkx基因)DNA甲基化低水平、表达水平升高;通过对候选基因的KEGG分析显示,Wnt信号通路中筛选出的新型候选基因与NTDs形成有关[23]。
在经典的Wnt信号通路中,Wnt2b是一种典型的Wnt基因,在原肠胚形成期、神经胚形成期及大脑皮质形成期,Wnt2b特定时空表达发挥重要功能[24]。Wnt7b通过PCP信号通路发挥作用并调控海马神经元的树突发育[25]。Wnt基因的异常表观遗传调控显示[24]:Wnt2b、Wnt7b启动子区发现异常组蛋白修饰,且在富含组蛋白修饰区域,Wnt2b、Wnt7b的DNA甲基化水平异常。Wnt2b、Wnt7b表达水平上调导致过多的信号和神经发育紊乱,Wnt基因异常DNA甲基化可能参与NTDs的形成。
神经管闭合依赖几种机制的联合作用:神经盘的汇聚延伸、神经上皮凋亡、神经嵴细胞迁移、增殖、分化,这些生物过程和形态学事件受多种信号通路及神经发育因子的共同作用。
(1) AKT2基因 AKT2是一种丝氨酸激酶。AKT2基因DNA高甲基化使易患NTDs的风险显著增加[16],异常的AKT2基因DNA甲基化引起基因表达改变,诱导细胞凋亡、增殖、分化,是NTDs发生的致病机制之一,AKT2基因DNA异常甲基化与NTDs发生的确切机制有待进一步阐明。
(2) HOXB7基因 HOX基因在神经管发育中起重要作用,以时空和共线方式被调控。一项甲基化水平病例-对照研究显示[26],HOXB7基因DNA低甲基化是脊髓脊膜膨出的危险因素。基因本体分析显示:候选基因通过神经元投射、突触处理、细胞投射等不同的通路共同起作用,候选基因DNA低甲基化及多种因素复合作用导致NTDs发生。
(3) TFAP2A基因 围孕期叶酸与神经嵴调节的DNA甲基化修饰研究[27]显示:叶酸摄入量减少与甲基化高水平有关,导致某些基因位点表达水平降低,其中TFAP2A基因是调节神经嵴发育的基因。进一步研究发现位于神经-面发育基因(TFAP2A和OTX2)启动子区附近的两个位点DNA甲基化水平升高,并揭示这两个基因参与神经嵴发育和颅面出生缺陷。
(4) Trim26、GNAS基因 人类胎儿脊柱裂与胎盘DNA甲基化研究[28]显示,在脊柱裂胎儿的胎盘中,Trim26、GNAS基因转录调控区保持低甲基化状态,通过基因本体分析发现有些基因直接参与神经系统发育和神经递质运输的调控;在这些基因中,许多是转录因子,参与神经发育调节,如:FEZF1、LBX1、VAX1、LMX1A。FEZF1调节神经元迁移,对胚胎神经管形成至关重要。胎盘DNA甲基化影响它的基因表达,导致胎儿发育畸形;GANS是重要的信号分子,介导许多信号通路,GANS蛋白表达受DNA甲基化调控,因此GANS基因的DNA甲基化水平影响NTDs的发生。
(5) Pax3基因 Pax3表达于神经上皮,是神经管闭合所需的基因,研究报道[29]在胚胎神经上皮和神经嵴诱导前,神经胚形成期Dnmt3b介导的Pax3基因的CpGs发生高甲基化,使Pax3基因表达沉默。
多数印迹基因的作用机制尚不清楚,然而几乎都与DNA甲基化型的异常相关。迄今发现的印迹基因有80多个,其中IGF2/H19模式与胎儿的生长发育和胎盘的功能密切相关,对于胚胎发育的基因表达调控非常关键。胰岛素样生长因子2(IGF2)是一种父源等位基因表达的促有丝分裂的生长因子[30],在胎儿和胎盘发育中起关键作用[31],与IGF1受体结合在细胞表面,IGF2蛋白激活细胞内多种信号瀑布,促进细胞生长[32]。而H19呈母源等位基因表达模式,并不翻译为蛋白质[33]。IGF2下游的DMR是一个印迹调控区,对IGF2和H19进行交互式的印迹调节。在NTDs发生的表观机制中,IGF2/H19的异常甲基化研究引起了学者的兴趣,但IGF2/H19甲基化在NTDs中的变异情况尚不统一。脊柱裂患儿的H19甲基化研究表明[21],荷兰和德克萨斯州两种人群均未发现该基因甲基化与脊柱裂的相关性,而一项国内研究表明[34],在NTDs病例组中,H19 DMR1的DNA甲基化水平明显升高。二者的结果之所以不同是因为Liu等[34]研究的是该基因上游5kb区域,而前者研究的是基因300~700 bp区域。随后H19DNA甲基化研究则表明[35],在人类NTDs中,H19基因DMR1呈高甲基化型,并且H19基因DNA高甲基化使IGF2表达上调,但IGF2基因DMR0甲基化水平无变化,因此推测IGF2表达水平升高可能不是因为IGF2 DMR0的甲基化型,而是受H19和IGF2的双重调控,异常基因组印迹可能是NTDs的致病因素。此外,IGF2甲基化研究[36]发现DMR0高甲基化是NTDs发生的危险因素,且具有性别依赖性,女性NTDs患儿DNA甲基化水平较高,表明女性胎儿的正常发育可能对甲基化修饰更敏感。因此NTDs患儿脑组织中IGF2 DMR0甲基化水平升高与NTDs风险增加有关。DMR0的亚组分析显示脊柱裂和无脑畸形患儿的甲基化水平升高。基于DMR0的高甲基化的NTDs风险评估提示高甲基化与NTDs风险增加相关[36]。脑组织中的DNA高甲基化降低IGF2的表达,检测肺和胎盘组织发现该基因DMR0甲基化水平均未升高,则提示IGF2基因DNA甲基化存在组织特异性。
构成核小体的组蛋白氨基端可以被多种酶进行修饰,如甲基化、乙酰化、泛素化、磷酸化等,组蛋白修饰可改变DNA-组蛋白的相互作用,使局部染色质构型发生改变。甲基化和乙酰化是最常见的修饰方式。组蛋白修饰主要发生在核心组蛋白H3和H4尾巴的精氨酸和赖氨酸残基上。维甲酸诱导的NTDs模型鼠的研究[24]显示Wnt2b、Wnt7b基因启动子区组蛋白H3第4位赖氨酸残基乙酰化(H3K4ac)修饰丰度上调,组蛋白H3第27位赖氨酸残基三甲基化(H3K27me3)修饰丰度下调,组蛋白H3第4位赖氨酸残基三甲基化(H3K4me3)修饰丰度未检测到;但在人类NTDs,H3K4ac修饰丰度下调,并且在人类脊髓样本发现Wnt2b、Wnt7b富含GC启动子区H3K4ac和H3K27me3修饰是丰富的。此外,还提示组蛋白修饰丰富的区域,Wnt2b、Wnt7b的DNA甲基化型异常,呈低甲基化型,说明组蛋白修饰和DNA甲基化之间相互影响。Wnt2b、Wnt7b基因启动子区H3K4ac、H3K4me3修饰与基因激活相关,H3K27me3修饰有助于基因沉默,且与发育调控基因的时空抑制相关。这种异常组蛋白修饰参与调控基因表达,在神经发育早期开始起作用,并具有时空特异性。有关启动子区的组蛋白修饰证据表明[35],组蛋白修饰调控的染色质状态与NTDs高度相关,H3K27me3发现在失活的启动子,而组蛋白乙酰化增强基因的表达。也有研究[37]揭示组蛋白H3第9位赖氨酸残基三甲基化(H3K9me3)修饰对细胞分化和胚胎形成必不可少,H3K9me3修饰缺失导致H3K9乙酰化修饰增多。
小分子核糖核酸(mircoRNA,miR)是约由20~25个核苷酸构成的单链非编码小RNA分子[38],主要表现为内源性的基因调节子,通过与3’-UTR区域结合,使靶向mRNA在转录后降解,负向调节基因表达[39]。mircoRNA作为转录后调节子,参与多种生物过程,包括神经系统发育及神经管发育[40]。miR-124a是中枢神经系统中表达最丰富的mircoRNA之一,在哺乳动物早期胚胎表达增加,胚胎发育晚期达到顶峰。鼠科动物中枢神经系统分化时,miR-124a表达上调可加速神经管发育。研究发现[39],miR-124a在脊柱裂小鼠脊髓中表达下调,并且miR-124a启动子区高甲基化的比例与它的表达水平呈负相关。维甲酸诱导的脊柱裂小鼠模型发现[41],miR-124a在有缺陷的脊髓中表达下调,miR-124a介导的染色质重塑调节机制可能参与脊柱裂的神经管缺损的发生,提示miR-124a表达紊乱是神经胚形成期发育缺陷的主要原因。此外,miR-302/367在神经胚形成期协调基因表达和神经前体细胞行为,敲除miR-302/367可导致神经前体细胞增殖和分化增加、早期胚胎致死和开放性的NTDs[42]。
人类无脑畸形胎儿脑组织中miR-149明显下调,miR-22、miR-125a表达上调,通过靶向基因的分子功能和生物过程分析表明,microRNA失调是无脑畸形发生的病因机制之一[40]。此外,利用microRNA芯片和qRT-PCR分析筛查NTDs孕妇血清中microRNA表达水平,并通过ROC曲线对血清microRNA进行诊断标志物评估,认为孕妇血清microRNA可能成为NTDs早期诊断的标志物[43]。
神经管闭合失败,对神经系统发育造成严重的影响,造成死胎、流产,或出生后严重的身体残疾,明确NTDs发病机制是预防其发生的关键,才能从根本上降低发生率,提高出生人口素质。DNA甲基化,组蛋白修饰,microRNA调节等表观遗传修饰异常参与调节神经管发育基因的表达水平改变,引起神经管闭合的多种生物过程和形态学事件不协调,但表观遗传修饰在NTDs形成中的具体机制尚不十分明确。因此,表观遗传修饰在NTDs病因中的作用有待进一步研究,有望为NTDs临床筛查提供理论依据。
无

























