
成骨不全(osteogenesis imperfecta, OI)是一种以骨脆性增加为特征的疾病,由COL1A1/COL1A2基因突变引起,根据患儿临床体征和组织病理学特性可将其分为Ⅰ~Ⅳ型,但Ⅴ型OI患儿中未发现Ⅰ型胶原基因突变。Ⅴ型OI患儿具有OI的共同特征:如多次非暴力性骨折、身材矮小、骨骼畸形等,但蓝巩膜发生概率较小,大多没有牙质形成不全和听力障碍。影像学表现以尺桡骨和/或胫腓骨骨间膜钙化、桡骨头脱位、增生性骨痂为特征。致病突变位于干扰素诱导跨膜蛋白5(interferon-induced transmembrane protein 5,IFITM5)编码基因的5'-非翻译区(5'-UTR),一个碱基C转换成T(c.-14C>T)。IFITM5基因在OI发病中的作用机制,已成为研究热点。本文将从Ⅴ型OI的临床表现和发病机制方面予以综述,旨在提高人们对该病的认识及诊断。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
成骨不全(osteogenesis imperfecta, OI)又称脆骨病,主要累及运动系统的结缔组织,亦可累及眼、耳、皮肤等。以全身骨骼脆弱、易骨折、骨骼畸形为主要特征。发病率在1/25 000至1/10 000[1]。1979年,Sillence等[2]根据临床表型将OI划分为4种亚型。但随着对该病认识的不断深入,越来越多的亚型被发现,有报道OI的分型已增加至15种[3]。分类标准从临床表型扩展到基因型,但临床表型和基因型间的相关性难以界定。2015年,国际骨骼体质障碍命名委员会(International Nomenclature Committee for Constitutional Disorders of the Skeleton)提出回归到Van Dijk和Sillence(2014)[4]提出的临床5个亚型分类,如表1所示。

OI分类命名和相关遗传方式[5]
OI分类命名和相关遗传方式[5]
| 分型 | 特点 | 遗传方式 | 备注 |
|---|---|---|---|
| Ⅰ型 | 无畸形型 | AD | 伴蓝巩膜 |
| Ⅱ型 | 围产期致命型 | AD,AR | |
| Ⅲ型 | 进行性畸形型 | AD,AR | |
| Ⅳ型 | 中度 | AD,AR | 巩膜一般正常 |
| Ⅴ型 | 伴骨间膜钙化和/或增生性骨痂 | AD |
注:AD,常染色体显性遗传;AR,常染色体隐性遗传
目前临床常用的分型方法是Sillence分型法,将OI分为Ⅰ~Ⅳ型,由COL1A1/COL1A2基因突变引起,但Ⅴ型OI患儿中未发现Ⅰ型胶原基因的突变,其临床表现及影像学特点也与Ⅰ~Ⅳ型存在差异。本文将从Ⅴ型OI的临床表现和发病机制方面予以综述,旨在提高人们对该病的认识及诊断。
2000年,Glorieux等[6]首先提出了Ⅴ型OI的概念。该病呈常染色体显性遗传,在OI患儿中所占比例不到5%[7]。Ⅴ型OI与其他型患儿的临床表现不同,一般表现为不同程度的前臂骨间膜钙化、桡骨头脱位和增生性骨痂[8,9,10,11,12]。影像学表现见图1。
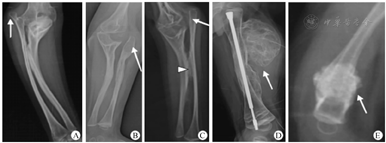
从目前的报道来看,前臂骨间膜钙化是几乎每个病例都存在的一个特征,多累及尺、桡骨骨间膜,也可累及胫腓骨骨间膜。尺、桡骨骨间膜钙化会导致前臂不同程度的旋前或旋后受限,并随着患儿年龄增长逐渐加重[6,8,9,10,11,12,13,14,15]。有研究报道骨间膜钙化在10岁前患儿中并不明显,最初仅表现为骨皮质增厚或内侧骨隆起[10,16]。但也有报道1岁就发生骨间膜钙化的患儿[17]。除了骨间膜,钙化还可以发生在肌肉起点、韧带甚至肌肉,比如腰方肌、内收肌、比目鱼肌在骨骼上附着点的钙化,胫腓韧带的钙化等[18]。
桡骨头脱位在许多患儿中都有发生,是肘关节畸形的原因之一[6,8,14,19]。桡骨头脱位首先表现为单侧,并随着患儿年龄增长而逐渐加重,后逐步发展至双侧。桡骨头脱位患儿大多有巨大尺骨鹰嘴和冠状突[16,17,20]。有学者认为,尺骨鹰嘴及冠状突异常增大可影响桡骨头发育,最终导致桡骨头脱位[21]。桡骨头脱位在Ⅰ、Ⅲ、Ⅳ型OI患儿中也偶有发生,发生率分别为1.5%、8.5%和6.5%;相比之下,桡骨头脱位在Ⅴ型OI患儿中似乎更为普遍,发生率可达约77%[20]。
高达65%的Ⅴ型OI患儿在骨折或外科手术后会出现增生性骨痂[11]。骨痂巨大呈圆形或椭圆形,增生性骨痂发生部位质地较硬、皮温较高,伴有疼痛、肿胀[20]。X线检查显示骨痂同股骨干界限分明,无骨皮质破坏,无骨膜反应,骨痂内密度低,巨大椭圆骨痂轮廓内可见不规则网格状分层形成;后期表现为巨大骨痂内密集钙化,密度同股骨干无明显差异,但股骨干界限仍可见,骨干皮质无破坏;术中可见骨痂内为大量脂肪组织,骨痂壁皮质菲薄,骨质脆弱[17]。
有学者认为,增生性骨痂是由于过度矿化所致,因为体外实验已经证实IFITM5和矿化之间存在明显正相关关系[22,23]。也有报道显示,增生性骨痂的发生与骨折或外科手术关系并不确切,这一现象的发生似乎是随机事件[20]。近年,有研究显示发生增生性骨痂患儿的红细胞沉降率(erythrocyte sedimentation rate,ESR)和C-反应蛋白(C-reactive protein,CRP)均有所增加,提示活动性炎症也在骨痂的形成过程中发挥了作用[20]。
Ⅴ型OI患儿除了以上3个比较公认的特征外,还有很多其他特征,如:关节挛缩、椎体压缩、脊柱侧弯和长骨弯曲。其中,脊柱侧弯多见于女性,长骨弯曲多见于胫骨和腓骨[20]。影像学特征如前臂干骺端致密带和骨组织学上网状的层状结构也是Ⅴ型OI特征[8,9]。
此外,有文献报道,部分Ⅴ型OI患儿有一些典型的面部特征,如:前额短而宽、眼距宽、鼻子扁平或者朝天鼻、嘴唇薄、下巴宽、双侧上颌后移、错颌畸形、下面部高度降低和多个牙齿缺失等[20,24,25]。这种面部特征提示,患儿可能存在颅面骨发育障碍。但目前这方面的文献报道比较少,还有待进一步研究。
Ⅰ~Ⅳ型OI患儿常见的临床表现,比如蓝巩膜、牙本质发育不全、听力受损等,在Ⅴ型OI患儿中比较少见[8,16]。随着更多研究的开展,Ⅴ型OI表型谱也在不断扩充,以前认为蓝巩膜、听力受损、牙本质发育不全等不是Ⅴ型OI的临床表现,现在陆续有研究发现这些特征也会在Ⅴ型OI患儿中出现[26]。
Ⅰ~Ⅳ型OI大多由COL1A1或COL1A2基因不同位点突变,使Ⅰ型胶原合成数量或结构缺陷所致[27]。但在Ⅴ型OI患儿中,未发现COL1A1或COL1A2基因突变。2012年,Cho等[8]对一个患有Ⅴ型OI的四代家族进行连锁分析和全外显子组测序,鉴定出一种杂合突变,该突变位于干扰素诱导跨膜蛋白5(interferon-induced transmembrane protein 5,IFITM5)编码基因的5'-非翻译区(5'-UTR),一个碱基C转换成T(c.-14C>T);并在来自3个家系的13例患儿和5例单一患儿中均发现了这种变异,而家族其他健康人均不存在这种变异。并且,在相同种族背景个体的200个无关正常染色体中也未发现这种变异,说明这种变异是引起Ⅴ型OI的致病突变。与此同时,Semler等[11]也通过全外显子组测序鉴定出这一突变。随后,更多文献报道在Ⅴ型OI患儿中检出了这种突变[9,10,15,16,21,24,28,29,30,31,32]。目前,该突变被认为是Ⅴ型OI致病突变。
IFITM5是干扰素诱导跨膜蛋白基因家族成员,位于11号染色体11p15.5,编码含132个氨基酸的蛋白。这种蛋白有1个跨膜结构域,N端位于细胞内,C端位于细胞外,在内质网或高尔基体中半胱氨酸残基被棕榈酰化,之后锚定在细胞膜上[33]。因为这种蛋白与其他IFITM家族成员簇集在一起且有IFITM样基因结构,所以被命名为IFITM5,但它没有干扰素反应元件,不能被干扰素诱导。再加上IFITM5只在人的骨和软骨中表达,因此也被称为骨限制性ifitm样蛋白(bone-restricted ifitm-like protein,BRIL)[8,23]。IFITM5在成骨细胞发育成熟及骨形成过程中有着重要作用。
IFITM5编码基因的5'-UTR的c.-14C>T突变导致Ⅴ型OI的机制尚不明确。该突变产生了一个新起始密码子,可能导致IFITM5的N端多出5个氨基酸(Met-Ala-Leu-Glu-Pro,MALEP)。但到目前为止,研究人员在Ⅴ型OI患儿骨骼样本中只检测到了MALEP-IFITM5转录产物,并未检测出MALEP-IFITM5蛋白[34]。体外实验中,携带IFITM5 c.-14C>T突变的HEK293细胞能够表达出一种比野生型IFITM5蛋白更长的蛋白质[11]。IFITM5敲除小鼠没有明显骨骼缺陷[35]。在IFITM5-/-小鼠中,长骨在出生时比IFITM5+/-小鼠短15%~25%,且它们有时会出现严重弯曲,这种症状到成年时部分会消失[36]。IFITM5-/-和IFITM5+/-小鼠胫骨骨形态参数也没有显著差异。这些数据表明,IFITM5蛋白缺失对骨量没有影响。2015年,Lietman等[37]建立了携带IFITM5 c.-14C>T突变的Ⅴ型OI小鼠模型,该小鼠模型在子宫内表现出矿化速度减慢,肋骨形成异常,长骨畸形和骨折。另一方面,在Ⅴ型OI小鼠模型中,Ⅰ型胶原是正常的,未出现其他类型中观察到的Ⅰ型胶原翻译后修饰。因此,在Ⅴ型OI小鼠模型中观察到的矿化速度降低和骨畸形被认为与Ⅰ型胶原结构改变无关。而Ⅴ型OI小鼠模型表现出围产期死亡,被认为是由于新生小鼠胸廓形成异常引起呼吸衰竭所致。由于Ⅴ型OI小鼠模型在围产期死亡,小鼠出生后是否会出现增生性骨痂和骨间膜钙化等特征便不得而知了。相反,过度表达野生型IFITM5的转基因小鼠生长正常,产前和产后发育正常,未见骨骼缺损。此外,这些转基因小鼠的骨形态参数与野生型小鼠没有差异。因此,IFITM5蛋白正常功能的增加对骨骼发育似乎也无影响。
有研究分离携带IFITM5 c.-14C>T突变患儿的成骨细胞进行原代培养,SERPINF1表现出过表达,从而导致其蛋白产物色素上皮衍生因子(pigment epithelium-derived factor,PEDF)表达水平升高[38]。由于外源性PEDF可以促进矿化作用,所以PEDF很可能参与了Ⅴ型OI患儿增生性骨痂的形成和骨间膜钙化[39,40]。但因为Ⅴ型OI小鼠模型在围产期死亡,PEDF是否参与了增生性骨痂的形成和骨间膜钙化都没能得到证实。如果PEDF参与了增生性骨痂的形成和骨间膜钙化,提示IFITM5 c.-14C>T突变所产生的MALEP-IFITM5蛋白能够通过调节SERPINF1表达,参与Ⅴ型OI的发病机制。
FK506结合蛋白(FK506-binding protein,FKBP)家族成员FKBP11(也称为FKBP19)是与小鼠IFITM5相互作用的伙伴分子[36],FKBP11是一种肽-脯氨酰基顺反异构酶。小鼠MALEP-IFITM5与FKBP11的相互作用与在野生型IFITM5中观察到的一致[23]。FKBP65与FKBP11同属FKBP家族,FKBP65(由FKBP10编码)基因突变可抑制赖氨酸末端肽羟基化,参与Ⅰ型胶原分子间交联形成,引起常染色体隐性遗传Bruck综合征和OI-XI[41]。FKBP10基因敲除小鼠表现出围产期死亡。这些敲除小鼠的颅骨胶原蛋白在赖氨酸末端肽分子间交联的形成中表现出不稳定性[41]。在IFITM5 c.-14C>T Ⅴ型OI小鼠模型中没有观察到这种对Ⅰ型胶原赖氨酸末端肽羟基化的抑制[37]。因此,可以排除MALEP-IFITM5通过抑制FKBP11功能来降低Ⅰ型胶原结构稳定性的情况。除FKBP11外,没有其他与IFITM5相互作用的蛋白被报道。与MALEP-IFITM5相互作用的蛋白质也未见报道。如能够鉴定出与MALEP-IFITM5蛋白特异性相互作用的蛋白,将有助于我们更好地理解IFITM5的功能以及造成Ⅴ型OI的原因。
IFITM5在成骨细胞矿化初期有特异性表达,体外实验中,成骨细胞中IFITM5过表达促进矿化;反之,减弱IFITM5表达则抑制矿化。因此,IFITM5被认为是一个矿化作用的正向调节因子。然而,IFITM5基因敲除小鼠(IFITM5-/-)在成骨过程中未显示任何明显异常。这些结果共同表明IFITM5在体内并非矿化的正向调节因子[23]。说明在体内可能存在不同的矿化作用机制。IFITM5基因敲除小鼠和IFITM5转基因小鼠没有明显骨骼异常,这提示我们,对于正常骨组织,IFITM5起到了抑制矿化的作用,Ⅴ型OI患儿中所观察到的增生性骨痂和骨间膜钙化可能是由于IFITM5抑制作用被解除所致。增生性骨痂是由于骨组织矿化过程中IFITM5抑制作用被解除,骨间膜钙化是由于在通常不骨化位置IFITM5抑制作用被解除。
虽然在Ⅴ型OI患儿中未发现Ⅰ型胶原基因的突变,但Reich等[22]分离Ⅴ型OI患儿成骨细胞进行原代培养发现,COL1A1基因表达显著降低,分泌型Ⅰ型胶原结构也出现异常。Ⅴ型OI患儿表现出的骨脆弱可能与Ⅰ型胶原异常有关,但MALEP-IFITM5影响分泌型Ⅰ型胶原水平和结构的机制尚不清楚。近年来,有学者在Ⅴ型OI患儿中发现了与FKBP10和PLOD2突变相关的重叠表型-关节挛缩[20,42],FKBP10和PLOD2参与Ⅰ型前胶原蛋白翻译后修饰、加工、折叠、分泌和交联过程。这提示我们,不同的OI致病基因与临床谱的重叠可能预示着这些基因之间存在着未知的潜在联系。
目前已经明确Ⅴ型OI是由IFITM5基因5'-UTR的c.-14C>T突变所致。但是IFITM5在骨形成机制中的作用、IFITM5功能在体内的调控机制以及MALEP-IFITM5对这些机制的影响都有待进一步阐明。IFITM5基因检测,将对患儿的产前诊断、遗传咨询、发病机制研究及将来基因治疗起到重要的促进作用。
所有作者均声明不存在利益冲突

























