
Beckwith-Wiedemann综合征(Beckwith-Wiedemann syndrome,BWS)是由位于11号染色体15.5区域印迹基因簇表达异常相关的一种罕见疾病。临床表现以腹壁缺损、巨舌和巨大儿为主,部分患儿可见耳褶皱及切迹、内脏肥大、新生儿低血糖、面部火焰状红斑等表现,易罹患胚胎性肿瘤且以肾母细胞瘤最为常见。本文通过对最新文献的复习总结,对BWS的基因结构、诊断流程、基因型和表现型、产前诊断和治疗管理进行综述。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
Beckwith-Wiedemann综合征(Beckwith-Wiedemann syndrome,BWS)又称脐膨出-巨舌-巨体综合征,是一种先天性过度生长疾病,最早在1955年被发现。Beckwith[1]和Wiedemann[2]分别于1963年和1964年报道此病,由此被命名为Beckwith-Wiedemann综合征。BWS发病率约为1∶13 700,无种族特异性,男女比例约为1∶1[3]。BWS被认为是一种临床综合征,主要表现有巨舌、腹壁缺损、偏侧增生、肾母细胞瘤及持续性低血糖(>1周)等[4,5],次要表现有巨大儿、面部火焰状红斑、羊水过多、耳褶皱及切迹、短暂性低血糖(<1周)、神经母细胞瘤、肝母细胞瘤、内脏肥大、腹直肌分离、脐疝等[4,5]。有3个主要表现或2个主要表现加1个次要表现的患儿可临床诊断BWS。本文通过对最新文献的综述,了解BWS的基因结构,归纳BWS诊断过程,描述BWS基因型和表现型关系,回顾BWS产前表现,探讨BWS的治疗和管理,以提高儿科医生对BWS的认识。
BWS由染色体11p15.5母源或父源性印记基因表达缺陷所致,印记中心为DMR(IC):H19/IGF2 DMR(H19-DMR/IC1/ICR1)和KCNQ1OT1:TSS DMR(KvDMR/LIT1-DMR/IC2/ICR2)(图1)[6]。父源等位基因在IC1上甲基化,母源等位基因在IC2上甲基化,可通过甲基化多重连接探针扩增技术(MS-MLPS)检测[7]。BWS具有复杂的基因型,80%BWS患儿印迹基因簇异常,以甲基化异常最常见,包括:①母源性IC2去甲基化(LOM)占50%;②父源性IC1获得甲基化(GOM)占5%~10%[8]。还可能的印迹基因簇异常包括:①父源性单亲二倍体(UPD)占20%;②CDKN1C基因突变占散发性BWS的5%和家族性BWS的40%;③11p15.5重复、倒置、微缺失和微重复(CNVs)等基因改变占<5%[8]。还有约20%BWS无明确分子诊断[5]。
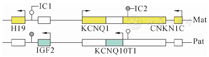
注:矩形区为印迹区调控的基因,父源等位基因在IC1上甲基化(灰色),母源等位基因在IC2上甲基化
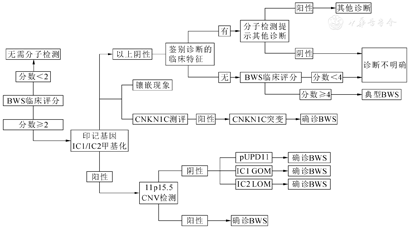
BWS的诊断主要根据临床表现。欧洲最新的BWS共识提出诊断BWS的分子诊断流程详见图2[4]。以主要表现为2分,次要表现为1分,总分<2分排除BWS,≥2分者行甲基化检测,≥4分可临床诊断BWS[4]。甲基化异常阳性者,进一步检测IC2LOM、IC1GOM、UPD、CNV[4,5]。甲基化异常阴性者,可能的原因和处理方式包括:①CDKN1C基因突变失功能;②送检其他组织排除体细胞镶嵌现象的干扰;③鉴别其他具有过度生长表现的疾病,比如Simpson-Golabi-Behmel综合征、Costello综合征、Perlman综合征、Maroteaux-Lamy综合征等[4,5,9]。体细胞镶嵌现象或染色体重排均可能导致检测结果呈阴性,故基因检测结果阴性不能排除BWS诊断[4,5]。
BWS散发病例约占85%,家族史阳性患儿约占10%~15%,属常染色体显性遗传。家族史阳性、基因检测确诊BWS者,其同胞和子代有患病倾向[10]。患病风险与缺陷基因的大小、位点、调控元件及亲本来源相关[4]。母源染色体CDKN1C致病性改变,其后代有50%的患病风险。重视CNVs对BWS家族遗传的重要性,涉及任一印迹区域的微缺失始终发生在母源染色体上,而微重复始终出现在父源染色体上[10]。明确基因型对BWS患儿及其家属的遗传咨询至关重要。
BWS以IC2LOM、IC1GOM、UPD及CDKN1C四个分子亚群为主,基因型与其表现型、患癌症风险有一定相关性[3,4,7,11,12]。其中,IC1GOM多表现为巨大儿、内脏肥大;IC2LOM、CDKN1C倾向出生后过度生长;UPD以偏侧增生更常见[4,5,13]。腹壁缺损多见于IC2LOM、CDKN1C,脐膨出以CDKN1C为主[13]。巨舌多与IC1GOM、IC2LOM相关[5,13]。IC1GOM、UPD罹患癌症风险高,尤其是肾母细胞瘤;而IC2LOM、CDKN1C患癌风险较低[5,12,13]。肝母细胞瘤则几乎仅见于UPD[12,13]。BWS各基因型间的临床表现存在重叠,了解BWS基因型和表现型的相关性有助于临床治疗、肿瘤筛查和并发症随访[3,4,7,11,12]。
BWS通常在出生后诊断,但家族史阳性或产检超声提示BWS者,建议行产前筛查[4,14]。William等[15]认为产前超声观察到2个主要表现或1个主要和2个次要表现即可诊断BWS。随着对疾病的认识和产检经验的积累,BWS的产检表现还有肾囊肿、额鼻角异常、头围四肢发育不协调、腭裂、蛛网膜下腔增宽、脐带偏长、单脐动脉等[7,10,16]。孕18~20周产检时重点观察胎儿腹部、颅面部及实质性器官大小,孕25~32周可确诊[3]。Shieh等[6]认为孕前期产检最常见的特征为小型脐膨出,巨大儿、巨舌、内脏肥大多在孕后期发现。约10%~20%产检发现孤立脐膨出的患儿最终被诊断为BWS[4,14]。产前生化指标中,孕早期hCG升高、孕中期AFP升高与BWS有关[4,16]。孕妇在孕期可能出现妊娠期糖尿病、妊娠期高血压、子痫、HELLP综合征及阴道出血[16]。
BWS的产前超声检出率为64.1%,且随时间进展无明显提高,32.7%的BWS患儿产检过程中未发现任何异常[14]。受胎儿体位和活动的影响,超声观察巨舌受限时行MRI检查。产前基因检测在孕早期可采集绒毛膜,孕后期可采集羊水细胞[17]。不仅要考虑镶嵌现象的干扰和样本培养过程中甲基化异常造成假阴性,还受技术水平及伦理问题限制[4,17]。
BWS的产前诊断为围产期管理、肿瘤早期筛查和长期监测提供了可能[14]。羊水过多、巨大儿、子痫和妊娠期糖尿病等妊娠合并症均可能引发早产。文献报道BWS的早产率在28.9%~53.0%[14,16,18]。巨大儿经阴道分娩可能发生肩难产、出生窒息、臂丛损伤甚至心脏骤停,故更倾向于剖宫产[19]。产前考虑BWS后规律产检及分娩应转至有能力处理BWS急性并发症(巨舌致气道阻塞、严重新生儿低血糖、腹壁缺损修补手术)的医学中心进行[3,4,5,7]。
90%的BWS患儿合并巨舌[20,21]。巨舌引起的严重气道阻塞导致的呼吸困难可通过行气管切开缓解[20]。随着舌生长速度下降和下颌骨生长,部分巨舌可自行缓解,但仍有约40%的患儿最终需行减舌手术[4,20]。以喂养困难为主,阻塞性睡眠呼吸暂停综合征,持续流口水,发音困难,口面部结构改变,由外表、语言等造成的社会心理问题均可作为减舌手术指征[4,22]。睡眠呼吸紊乱可先尝试体位疗法、持续气道正压等保守治疗方式[21]。2~3岁前接受手术效果更佳,通常在1岁后行以前楔形切除术为主的舌减容手术,必要时可联合行扁桃体腺样体切除术、舌系带切除术等以达到更好的手术效果[20,21]。术中需注意保护舌神经及血管,术后需关注出血感染、伤口裂开、舌头水肿堵塞气道、舌组织再生及舌体不对称等并发症。长期随访显示,大多数患儿预后良好,进食困难、外观及语言能力得到改善,口水减少,味觉不受损[22,23]。
60%~77%的BWS患儿合并腹壁缺损[6]。但腹壁缺损的程度存在差异,主要表现有脐膨出、脐疝、腹直肌分离等[13]。Kumar等[24]认为脐膨出者均需行BWS筛查。75%~80%的BWS患儿可在产检时发现脐膨出,诊断脐膨出的胎儿建议行染色体核型分析[25]。脐膨出患儿出生后应仔细评估心肺状况,必要时行气管插管,留置胃肠减压管,纱布覆盖保护包膜完整性,建立静脉通路防止体液流失而低血容量性休克,可预防性使用抗生素[25]。需注意的是,无论脐膨出大小均有包膜破裂的风险[26]。小型脐膨出可行一期修补术,切除囊膜,结扎脐血管,将膨出内容物回纳后分层缝合腹壁缺损。对于脏器还纳困难可能造成患儿呼吸与循环障碍,手术以减张为目的,行皮肤覆盖缝合或借助生物敷料形成腹壁疝,二期行腹壁疝修补术。巨型脐膨出患儿可借助Silo袋,袋顶悬挂于暖床利用重力使脏器逐步回纳腹腔,再分期缝合腹腔[26]。也有医学中心选择磺胺嘧啶银或聚维酮碘涂抹,以促进上皮化延期修补腹壁或采用负压吸引法治疗巨型脐膨出[27,28]。脐疝腹壁缺损程度小,无症状者建议4岁至成年前完善修补术[29]。术后可能伴随进食困难、发育落后、胃食管反流和粘连性肠梗阻等并发症[30]。
30%~60%的BWS患儿合并新生儿低血糖[12,13]。新生儿低血糖可引起震颤、阵发性青紫、呼吸暂停或急促、肌张力低下、反应差以及嗜睡甚至惊厥等症状。出生6 h内连续2次血糖水平<2.75 mmol/L即可诊断。BWS低血糖多为暂时性低血糖,立刻予10%葡萄糖溶液2 ml/kg静脉推注,静脉通路建立后以6~8 mg·kg-1·min-1维持血糖水平>3.9 mmol/L,一般生后3 d可至正常水平(3.5~5.5 mmol/L)[31]。低血糖发病越早、水平越低、持续时间越长,越易造成中枢神经系统的永久性损害[31]。超过一周的持续性低血糖,可采取口服氯甲苯噻嗪(5~15 mg·kg-1·d-1,2次/d),皮下注射奥曲肽(2~20 mg·kg-1·d-1,每6~8 h/次),持续静脉滴注胰高血糖素(1 mg/d)等药物支持治疗,必要时可行胰腺次全切除术,切除范围根据PET/CT及术中活检结果决定[32,33]。术后胰岛素依赖型糖尿病发病率会随着随访时间的延长长期而增加[34]。新型药物如西罗莫司、胰高血糖素样肽-1受体(GLP1R)拮抗剂正逐步应用于新生儿低血糖的治疗。
43%~65%的BWS患儿存在过度生长[12,13]。BWS患儿的体重和身长均显著高于正常水平,增长水平按IC1GOM、UPD、IC2LOM、CDKN1C递减,与分子机制引发的表现型变化保持一致[9,18]。IC1GOM可引起胰岛素样生长因子IGF2双等位基因表达和抑癌基因H19表达下降,UPD可引起IGF2双等位基因表达和细胞周期抑制基因CDKN1C、H19表达下降,IC2LOM可引起CDKN1C基因表达下降。IGF2表达程度与胎儿增生程度有关,CDKN1C表达下降和突变导致临床出现过度生长、器官增大。BWS过度生长在儿童期后期逐渐减缓,可详细记录身高、骨龄、体重及四肢长度变化,绘制特定的BWS生长发育图表[18,35]。目前尚无BWS过度生长干预治疗的研究数据。偏侧增生以肌肉体积不对成增加及骨骼不对称为主[11]。上肢不对称过度生长通常不需要干预,双下肢长度差异<2 cm可穿增高鞋垫,>2 cm可采取骨骺固定术,避免继发性脊柱侧弯和反复背部疼痛[35,36]。
BWS胚胎肿瘤总体罹患率为8%[37]。最常见的肿瘤类型为肾母细胞肿瘤(52%),肝母细胞瘤(14%)、神经母细胞瘤(10%)、横纹肌肉瘤(5%)和肾上腺肿瘤(3%)[12]。2岁前患癌风险最高,在青春期之前逐渐下降至一般人群患癌风险水平,目前没有报道成年后有增加趋势[4]。BWS患儿的患癌风险存在基因型差异,比如IC1GOM多患Wilms肿瘤,IC2LOM多患肝母细胞瘤,CKDN1C突变多患神经母细胞瘤[12,37,38]。建议超声监测BWS相关肿瘤直至7岁[4,38]。肾母细胞瘤首选B型超声,肝母细胞瘤筛查通常结合B型超声和血AFP水平。但AFP用于早期诊断肝母细胞瘤存在争议,部分学者认为血清AFP波动大不推荐用于筛查,也有学者认为分子诊断为UPD的BWS监测AFP水平是有意义的[4,5,37]。神经母细胞瘤监测结合B型超声和尿高香草酸/香草扁桃酸(HVA/VMA)。此外,癌症可能是BWS的首要显性表现。MacFarland等[38]报道了12例以肾母细胞瘤为首要表现而最终确诊的BWS。长期随访的目的是早期发现肿瘤并干预,可根据BWS分子诊断结果制定个体化肿瘤监测方案,以便早发现、早诊断、早治疗同时避免不必要的心理压力和经济损失。
BWS患儿的认知及神经发育受多种因素影响。新生儿低血糖可对中枢神经造成损害,甚至造成不可逆的脑损伤[31,32]。巨大儿难产史缺血缺氧脑病可对大脑发育同样造成影响[36]。有研究将贝利婴儿发展量表用于调查脐膨出患儿并发症及预后情况,发现其认知、运动和语言的综合得分均落后于同龄人,并认为术前腹壁缺损大小、肺发育不全,围手术期血气、呼吸机运用及术后感染、肺动脉高压、延迟喂养营养落后均可对神经发育造成影响[36,39]。在接受教育后,患儿的认知能力会先于运动能力逐渐改善,建议随访神经系统发育情况和尽早开展康复训练,争取获得与同龄人相似的健康状况和生活质量[39]。Kent等[40]首次将长处与困难问卷(SDQ)问卷表运用到BWS患儿神经行为发育研究中,发现其在情绪及同龄人相处的测量中得分偏高,可能与外貌差异和印迹基因差异表达相关,部分患儿经精神疾病诊断与统计手册(DSM)确诊自闭症[39]。目前BWS患儿长期认知和神经发育仍需进一步研究。
13%~20%的BWS患儿合并先天性心脏病[4]。出生后常规行先天性心脏病筛查,管理原则与未合并其他畸形的先天性心脏病患儿一致。动脉导管未闭、卵圆孔未闭、房间隔缺损或室间隔缺损等小缺损可心动超声随访待其自行愈合,合并严重先天性心脏病者需手术矫正,建议行3~5年/次超声心动图随访[4,41]。28%~61%的BWS患儿合并良性泌尿系统疾病[4]。出生后常规筛查泌尿系统畸形,关注尿钙水平,腹部超声关注肾肥大、肾积水、肾结石及肾囊肿可能,严重膀胱输尿管反流可引起反复尿路感染和肾功能损伤[42]。同样建议行3~5年/次泌尿系统超声随访及肾功能检查[41]。
结合临床表现和基因检测,BWS诊断主要在出生后。产前也可以实现诊断,常见的超声表现有巨大儿、脐膨出、巨舌及羊水过多[15]。新生儿阶段是BWS存活的关键时期,一年生存率高达90%[14,29]。临床医生需要对BWS加强认识并及时诊断,尽早开始BWS易感肿瘤的筛查和防治,关注远期认知及神经发育情况,诊治过程多学科合作长期随访,如NICU、新生儿外科、心脏外科、矫形外科及临床心理科[4,5]。目前关于BWS远期健康情况和晚期并发症的数据缺乏,建议规律随诊至16~18岁[4]。临床医生不仅要具备诊治BWS的能力,也要关注BWS患儿家庭面临的经济和社会心理压力,必要时应转诊至专业的遗传和心理咨询中心。
所有作者均声明不存在利益冲突

























