
探讨TBX20基因突变与先天性心脏病的关系,探索先天性心脏病发生的分子生物学机制。
收集2017年8月至2019年7月在山西省儿童医院心胸外科确诊的353例中国汉族先天性心脏病患儿的临床资料作为观察组,其中男206例,女147例;中位年龄为1岁9个月,年龄范围为0~13岁,以同期门诊体检健康儿童350例为对照组,提取基因组DNA。通过聚合酶链式反应(polymerase chain reaction,PCR)扩增编码TBX20基因的外显子以及外显子内含子交界区并进行测序分析。分别应用SIFT、PolyPhen2和Mutation Taster软件和PrDSM模型集成软件对发现的错义突变和同义突变进行致病性预测。
研究发现2个新错义突变,2个新同义突变。在1例完全性肺静脉异位引流合并房间隔缺损的患儿中发现新错义突变c.361A>T(I121F);在1例法洛氏四联症合并卵圆孔未闭和动脉导管未闭的患儿中发现新错义突变c.785C>T(T262M)。在1例房间隔缺损合并不完全性右束支传导阻滞的患儿中发现新同义突变c.666C>T(N222N);在1例法洛氏四联症合并卵圆孔未闭的患儿中发现新同义突变c.786G>C(T262T)。这些变异在对照组儿童中均未检测到。I121F、T262M、N222N、T262T均位于高度保守的T-box DNA结合结构域内。致病性预测表明除T262T致病可能较小外,I121F、T262M和N222N均为致病性突变,影响蛋白质功能。
TBX20基因突变在中国儿童先天性心脏病房间隔缺损、法洛氏四联症、完全性肺静脉异位引流的发病机制中可能发挥着重要作用。这一发现首次将TBX20基因突变与完全性肺静脉异位引流疾病联系起来,拓宽了TBX20基因突变的疾病谱。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
先天性心脏病(congenital heart disease,CHD),是指胚胎时期心脏发育过程中出现的结构性缺陷[1]。这些缺陷可能发生在心房和心室之间的间隔、心脏瓣膜或大动脉和静脉[2]。患有复杂CHD的婴儿大多在出生后1年内死亡,死胎中有10%是由CHD引起[3,4]。CHD发病率高,临床上意义重大,尽管很多学者也进行了相关研究,但先心病的发病机制仍不明确。参与转录调控、信号转导和染色质修饰的基因发生突变均可在心脏发育过程中引发其结构和功能紊乱,目前在先心病患儿中报道的基因有NKX2.5、GATA5、ZIC3和TBX20等[5,6,7,8]。TBX20是T-box转录因子家族的一个重要成员,在脊椎动物和无脊椎动物胚胎发育的心脏中均表达。TBX20位于第7号染色体(7p14.2)上,有8个外显子,其289-888核苷酸及相应氨基酸编码T-box DNA结合结构域,通过此功能结构域与特异DNA序列结合,从而调控转录。已有学者在CHD中进行了一些研究,发现TBX20在CHD的发生机制中可能发挥着重要作用[6, 9,10,11,12]。为进一步探讨TBX20与CHD之间的关系,本研究对353例中国汉族儿童CHD进行了TBX20基因突变检测,入选CHD畸形大部分为散发CHD,旨在为评估中国CHD儿童TBX20基因突变的发生率以及探讨基因型和表型的相关性提供依据。
收集2017年8月至2019年7月在山西省儿童医院心胸外科确诊的353例中国汉族CHD患儿的临床资料作为观察组,大部分为散发儿童,其中男206例,女147例;中位年龄为1岁9个月,年龄范围为0~13岁。353例中室间隔缺损150例,房间隔缺损63例(其中5例存在先心病家族病史),法洛氏四联症62例,完全性肺静脉异位引流17例,心内膜垫缺损16例,大动脉转位14例,肺动脉闭锁6例(其中1例存在先心病家族病史,此患儿的姐姐也患有肺动脉闭锁),肺动脉狭窄19例,右室双腔心4例,主肺动脉窗2例。所有患儿的心脏畸形均经过体格检查、彩色超声心动图及心脏外科手术或介入治疗后确诊,并排除心脏血管以外的其他畸形和遗传代谢性疾病,如Noonan,DiGeorge,Holt-Oram,Marfan,Alagille和Char等综合征。此外,对出现TBX20突变的患儿父母、兄弟姐妹也进行外周血DNA突变检测。选取350名无血缘关系、性别匹配且无明显遗传和出生缺陷等条件的健康儿童作为对照组。本研究通过山西省儿童医院医院伦理委员会批准(批件号IRB-KY-2017)。所有参与本研究的患儿法定监护人均知情同意。
采集观察组和健康对照组儿童外周血3 ml置于枸橼酸钠(EDTA)抗凝管中,放置于-20 ℃冰箱中备用。提取外周血白细胞基因组DNA,应用紫外分光光度计进行质量检测,检测无误后将DNA样品置于-70 ℃冰箱中保存。
根据TBX20基因(Genbank基因序列号NM_001077653)编码T-box DNA结构域的氨基酸位于第2-6外显子[13],本研究应用Primer Premier5软件设计引物(表1),采用聚合酶链式反应(polymerase chain reaction,PCR)扩增编码区和外显子、内含子交界区。每一个反应体系为50 µl,包括基因组DNA模板2 µl,10xPCR Buffer(Mg2+Plus)5.0 µl,dNTPs 3.0 µl,上、下游引物(0.3 μM)各1.5 µl,TaqDNA聚合酶(5 U/µl)(TakaRa,大连宝生物公司)0.4 µl,去离子水加至50 µl,在美国PTC-200型自动扩增仪上进行反应。设置PCR反应条件为:首先95 ℃预变性5 min,95 ℃变性30 s、54~60 ℃退火30 s及72 ℃延伸45 s,终延伸72 ℃ 5 min,共36~40个循环。将PCR扩增产物进行电泳分析。

TBX20基因扩增引物序列
TBX20基因扩增引物序列
| 外显子 | 引物序列(5'-3') | 扩增片段长度(bp) | |
|---|---|---|---|
| 2 | 上游 | CCCAGGTAATTTTATACAGTGAT | 597 |
| 下游 | CTAGACATCCTGTAGCTCCTAAT | ||
| 3 | 上游 | GAGTCAGACCCTTTCCCTCC | 401 |
| 下游 | AGGCTTGGAATGCTCTCTTG | ||
| 4 | 上游 | CCCACTTATATATGGTTTATGTGTTCC | 333 |
| 下游 | AGATAGAAGGTGGGAAGGGG | ||
| 5 | 上游 | TTAAGGCTGGAAAGGGTAGAT | 541 |
| 下游 | AGGTGTGGGTGGGGAAGTG | ||
| 6 | 上游 | TTCCACCCTTCTCAGGACAC | 708 |
| 下游 | AGGCCTGCCTGATGTCTCT | ||
切取含有目的基因片段的凝胶条,按北京天根生化科技有限公司的琼脂糖凝胶DNA回收Kit说明书进行DNA回收操作。将纯化后的PCR扩增产物直接测序,若检测到的突变,则将该样本再进行反向测序,验证测序结果的可靠性。对测序结果不十分明确、可疑变异的序列c.785C>T,将PCR产物克隆到pGEM-T载体中,挑选多个重组子再分别测序进一步证实。
应用DNASTAR和Chromas软件观察结果,将测序结果与Genbank人TBX20基因组DNA序列进行比对,识别TBX20基因突变。如果发现存在TBX20突变,则检索PubMed数据库、SNP数据库及万方数据库以评估所发现基因突变的新颖性。
登录美国国立生物信息中心(National Center for Biotechnology Information,NCBI)数据库,获得人、黑猩猩、鼠、牛、狗和斑马鱼等多物种的TBX20蛋白质氨基酸序列,应用NCBI网站Homologene进行多序列对比分析并用DNAMAN软件作图,明确所改变氨基酸在进化上的保守性。
通过对353例CHD患儿的TBX20基因编码区进行测序分析,在2例CHD患儿中检测到2个错义突变(I121F和T262M),1例患有心上型完全性肺静脉异位引流合并房间隔缺损,另1例患有法洛氏四联症合并卵圆孔未闭和动脉导管未闭,在健康对照组中均未发现(表2)。突变I121F和T262M均位于TBX20 T-box DNA结构域内,前者位于第3外显子区,后者位于第4外显子区。突变I121F使第121位的异亮氨酸(Ile)变为苯丙氨酸(Phe),突变T262M使第262位的苏氨酸(Thr)变为蛋氨酸(Met)(图1)。


本研究新发现的先天性心脏病患儿TBX20错义突变
本研究新发现的先天性心脏病患儿TBX20错义突变
| 编码区序列变异 | 氨基酸改变 | 病例数(例) | 先心病类型 | 家族史 | 父母亲 | 同胞 |
|---|---|---|---|---|---|---|
| c. 361A>T | I121F | 1 | TAPVC+ASD | - | - | 无同胞 |
| c. 785C>T | T262M | 1 | TOF+PFO+PDA | - | - | - |
注:TAPVC,完全性肺静脉异位引流;ASD,房间隔缺损;TOF,法洛氏四联症;PFO,卵圆孔未闭;PDA,动脉导管未闭;"-"表示阴性
在2例CHD患儿中检测到2个杂合序列变异c.666C>T(N222N)和c.786G>C (T262T)(图2),它们均未造成氨基酸改变。2例患儿中的1例患有房间隔缺损合并不完全性右束支传导阻滞,另1例患有法洛氏四联症合并卵圆孔未闭,他们均无先心病家族史(表3)。这2个同义突变均位于TBX20 T-box DNA结合域内,在对照组中均未发现。

注:箭头示突变位置,c.666C>T:N222N;c.786G>C:T262T

本研究新发现的CHD患儿TBX20同义突变
本研究新发现的CHD患儿TBX20同义突变
| 核苷酸序列改变 | 氨基酸改变 | 病例数(例) | 先心病类型 | 家族史 |
|---|---|---|---|---|
| c. 666C>T | N222N | 1 | ASD+IRBBB | - |
| c. 786G>C | T262T | 1 | TOF+PFO | - |
注:ASD,房间隔缺损;IRBBB,不完全性右束支传导阻滞;TOF,法洛氏四联症;PFO,卵圆孔未闭;"-"表示阴性
蛋白质的结构和功能由氨基酸的性质决定,而有些氨基酸在物种进化过程中相对稳定,不易变化,并且这种氨基酸直接影响蛋白质的关键功能,这就是所谓的氨基酸的保守性。这类保守性氨基酸若发生突变将直接导致某种或几种疾病的发生。在临床诊疗中,如果提早检测出这类保守性氨基酸发生的变化,则有可能预测出某些特定疾病的发生,为疾病的早期诊疗与干预提供参考。本研究对新发现的TBX20基因突变位点编码的氨基酸进行不同物种(包括人类、黑猩猩、鼠、牛、狗、马、鸡和斑马鱼)序列比对,结果显示第121位异亮氨酸和第262位苏氨酸在物种进化上高度保守(图3)。

注:第121位的异亮氨酸(I)和第262位的苏氨酸(T)高度保守
经SIFT、PolyPhen2和Mutation Taster3个软件预测错义突变I121F和T262M均具有致病性并影响蛋白质功能。TBX20同义突变N222N和T262T经PrDSM集成预测软件分析,结果显示FATHMM-MKL和PrDSM软件预测提示N222N致病性可能大,T262T致病可能较小。
先天性心脏病为胚胎期心脏和/或大血管结构异常导致的先天性心脏畸形[16],其特征是心腔、间隔、瓣膜以及大血管的形态发生异常[17]。由于解剖结构及血流动力学特点不同,CHD有很多种类型,如动脉导管未闭(patent ductus arteriosus, PDA)、肺动脉狭窄、房间隔缺损(atrial septal defect, ASD)、室间隔缺损(ventricular septal defect, VSD)、法洛氏四联症(tetralogy of fallot, TOF)、Ebstein畸形、冠状动脉起源异常、大动脉转位、永存动脉干、右心室双出口(double outlet right ventricle, DORV)、主动脉缩窄和心内膜垫缺损等等[18]。CHD是小儿心血管疾病中的重要组成部分,在临床重大出生缺陷中占到很大比例,每1000名活产儿中约有4~50人发生CHD[19]。大约80%的CHD病因不明,目前普遍认为它们遵循着多因素遗传模式,多种基因之间相互影响,并与环境因素相互作用,从而提高CHD患病的可能性[20]。关于CHD遗传机制研究方面,随着新一代测序技术的改进和效率提高,可能将发现更多与CHD相关的基因变异[21]。在已发现的致病基因中有很大一部分编码心脏转录因子,如同源结构域转录因子,锌指转录因子和T-box转录因子等[11]。本研究则是围绕T-box转录因子TBX20对CHD的发病原因进行初步研究。
T-box转录因子由180个氨基酸组成,具有序列特异的T-box DNA结合域且高度保守。T-box家族的一些基因在人类遗传性疾病(包括综合征性出生缺陷、先天性心脏病、扩张型心肌病和心律失常)中的一些重要作用已经得到确认,TBX1亚家族(TBX1、TBX18和TBX20)和TBX2亚家族成员(TBX2、TBX3和TBX5)在心脏发育过程中至关重要的心原性谱系细胞中呈现出综合性时空细胞生物学表达模式[22]。TBX20是TBX1亚家族成员,由3.9kb的核苷酸序列组成,编码含有446个氨基酸的蛋白质[23]。一系列动物研究发现,在果蝇、斑马鱼、鸡的心脏发育过程中,Tbx20以一种非常保守的方式发挥着重要作用[24,25],可见在生物进化过程中Tbx20对于心脏发育有着不可或缺的价值,而在哺乳动物中的研究也证实了这一观点。Tbx20在小鼠心脏发育的整个过程中都有表达[23]。Tbx20基因敲除的小鼠表现出流出道及右心室发育不良[26]。在人类研究中,Kirk等[27]在2例患有ASD的患儿中发现2个TBX20突变,即错义突变I152M和无义突变Q195X,首次将转录因子TBX20突变与人类病理性心脏疾病关联起来。2012年Qiao等[28]首次提出TBX20基因启动区域的序列改变与CHD有关的假设,并且在265例VSD病例中发现新杂合突变g.4932G>A,使TBX20启动子活性降低,认为这可能会导致VSD的发生。2015年Monroy-Muñoz等[29]首次在TBX20转录激活区域内发现与ASD相关的错义突变Y309D、T370O和M395R,为后续研究提供了借鉴。Pan等[10]在DORV患儿中检测到TBX20错义突变R143W,并发现该突变会降低TBX20转录活性,他们首次将失功性TBX20突变与DORV联系起来。随后,有学者在扩张性心肌病患儿和鼠动物模型研究中发现,患儿和鼠的TBX20 mRNA表达水平均显著升高[30]。Zhou等[31]在115例散发的先天性特发性扩张性心肌病患儿中发现TBX20无义突变E143X,并在进一步功能研究中发现该突变导致产生截断蛋白,造成TBX20的转录活性消失,同时使TBX20与NKX2.5及与GATA4之间的协同作用丧失。目前TBX20基因突变位点研究及相关CHD类型文献总结见表4。
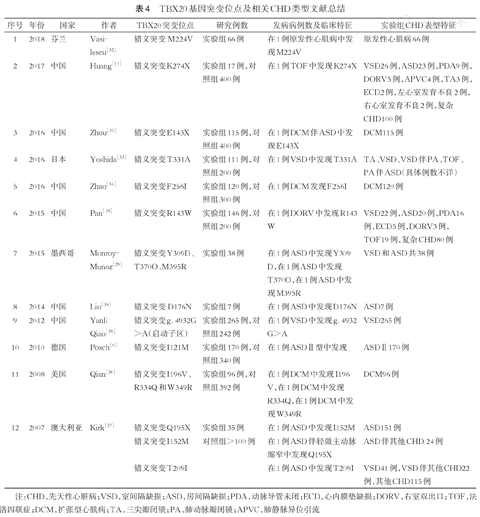
TBX20基因突变位点及相关CHD类型文献总结
TBX20基因突变位点及相关CHD类型文献总结
| 序号 | 年份 | 国家 | 作者 | TBX20突变位点 | 研究例数 | 发病病例数及临床特征 | 实验组CHD表型特征 |
|---|---|---|---|---|---|---|---|
| 1 | 2018 | 芬兰 | Vasilescu[32] | 错义突变M224V | 实验组66例 | 在1例原发性心肌病中发现M224V | 原发性心肌病66例 |
| 2 | 2017 | 中国 | Huang[11] | 错义突变K274X | 实验组17例,对照组400例 | 在1例TOF中发现K274X | VSD26例,ASD23例,PDA9例,DORV5例,APVC4例,TA3例,ECD2例,左心室发育不良2例,右心室发育不良2例,复杂CHD100例 |
| 3 | 2016 | 中国 | Zhou[31] | 错义突变E143X | 实验组115例,对照组400例 | 在1例DCM伴ASD中发现E143X | DCM115例 |
| 4 | 2016 | 日本 | Yoshida[33] | 错义突变T331A | 实验组111例,对照组200例 | 在1例VSD中发现T331A | TA、VSD、VSD伴PA、TOF、PA伴ASD(具体例数不详) |
| 5 | 2016 | 中国 | Zhao[34] | 错义突变F256I | 实验组120例,对照组300例 | 在1例DCM发现F256I | DCM120例 |
| 6 | 2015 | 中国 | Pan[10] | 错义突变R143W | 实验组146例,对照组200例 | 在1例DORV中发现R143 W | VSD22例,ASD20例,PDA16例,ECD5例,DORV3例,TOF19例,复杂CHD80例 |
| 7 | 2015 | 墨西哥 | Monroy-Muñoz[29] | 错义突变Y309D、T370O、M395R | 实验组38例 | 在1例ASD中发现Y309 D,在1例ASD中发现T370O,在1例ASD中发现M395R | VSD和ASD共38例 |
| 8 | 2014 | 中国 | Liu[35] | 错义突变D176N | 实验组7例 | 在1例ASD中发现D176N | ASD7例 |
| 9 | 2012 | 中国 | Yanli Qiao[28] | 错义突变g. 4932G>A(启动子区) | 实验组265例,对照组242例 | 在1例VSD中发现g. 4932 G>A | VSD265例 |
| 10 | 2010 | 德国 | Posch[6] | 错义突变I121M | 实验组170例,对照组340例 | 在1例ASDⅡ型中发现 | ASDⅡ170例 |
| 11 | 2008 | 美国 | Qian[36] | 错义突变I196V、R334Q和W349R | 实验组96例,对照组392例 | 在1例DCM中发现I196 V,在1例DCM中发现R334Q,在1例DCM中发现W349R | DCM96例 |
| 12 | 2007 | 澳大利亚 | Kirk[37] | 错义突变Q195X | 实验组35例 | 在1例ASD中发现I152M | ASD151例 |
| 错义突变I152M | 对照组>100例 | 在1例ASD伴轻微主动脉缩窄中发现Q195X | ASD伴其他CHD 24例 | ||||
| 错义突变T209I | 在1例ASD中发现T209I | VSD41例,VSD伴其他CHD22例,其他CHD115例 |
注:CHD,先天性心脏病;VSD,室间隔缺损;ASD,房间隔缺损;PDA,动脉导管未闭;ECD,心内膜垫缺损;DORV,右室双出口;TOF,法洛四联症;DCM,扩张型心肌病;TA,三尖瓣闭锁;PA,肺动脉瓣闭锁;APVC,肺静脉异位引流
本研究在4例先心病患儿中检测出2个新的错义突变I121F和T262M、2个同义突变N222N和T262T,它们在350名健康对照组中均未发现。这4个突变均位于高度保守的T-box DNA结合结构域内。通过对人和多种动物的TBX20氨基酸序列进行比对后发现,错义突变I121F和T262M中121位的异亮氨酸(I)和262位的苏氨酸(T),在人类、黑猩猩、牛、狗、鼠、马、鸡及斑马鱼中高度保守(图3)。这2个突变均使TBX20编码区的氨基酸发生改变,突变I121F使第121位的非极性、疏水性异亮氨基酸(I)被苯丙氨酸(F)取代,取代后的苯丙氨酸属于芳香基侧链,与之前不同,这一改变有可能造成蛋白质原有的空间结构及稳定性发生变化。突变T262M使第262位苏氨酸(T)变为蛋氨酸(M),这是从极性、中性氨基酸到非极性、疏水性氨基酸的改变,可能会影响蛋白质空间结构构象的变化,从而导致蛋白质原有功能的异常。
对于本研究新发现的2个同义突变N222N和T262T,传统观点曾认为这种突变没有造成氨基酸变化不改变蛋白产物成分,所以属于非病理性的。但在随后的许多研究中均发现同义突变会在某个或多个方面对蛋白质的结构和功能产生影响,比如对剪切调控元件的影响,导致可能产生新的可变剪切子,或者改变mRNA的翻译动力学从而影响蛋白质的空间构象,再者同义突变也可能影响活体内蛋白质的折叠进而影响蛋白质功能[38,39,40,41,42,43]。本研究将此次发现的TBX20错义突变和同义突变利用国际上流行的SIFT, PolyPhen2和Mutation Taster等多个软件分别进行了致病性预测分析,结果表明,除T262T致病可能较小外,I121F、T262M和N222N均为致病性突变,影响蛋白质功能。因此,这些特有的新发现的突变,尤其I121F、T262M和N222N,很可能与CHD发病机制相关或提高CHD的患病易感性,但要明确这些突变的具体作用本课题组将会在下一步的功能研究中深入探讨。
本研究发现突变I121F、T262M、N222N和T262T的4例患儿1例并发完全性肺静脉异位引流(TAPVC),2例并发TOF,其中1例同时合并PDA,另1例并发不完全性右束支传导阻滞,但4例同时都有ASD或者卵圆孔未闭(PFO)。这些CHD表型特征与Tbx20在鼠中的表达以及鼠模型研究中将Tbx20水平敲低后出现的效应基本一致[26]。
本研究首次在TAPVC患儿中检测到TBX20突变,第一次将TBX20突变与TAPVC联系起来,拓宽了TBX20基因突变的疾病谱,为CHD的发生机制提供了一点新思路。
所有作者均声明不存在利益冲突

























