
版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
缺血再灌注损伤包含缺血和再灌注两个过程,缺血性损伤由血流量阻断而使组织缺血缺氧所致,再灌注损伤则是缺血组织恢复血供过程所引起的缺血性组织损伤的进一步加重。肾脏缺血再灌注损伤在肾移植过程中不可避免,并对肾移植的成功率和移植后的肾脏功能恢复有重要影响。肾脏缺血再灌注损伤的机制异常复杂,其中涉及到钙超载导致的线粒体损伤、活性氧自由基(ROS)的作用、能量代谢障碍和多种细胞因子的参与等,因缺血再灌注损伤的发生机制尚未完全明确,预防和治疗肾脏缺血再灌注损伤仍缺乏有效的手段。因此,深入探讨肾脏缺血再灌注损伤的机制,对降低肾脏缺血再灌注损伤的发生率和加强保护性干预有重要意义。
瞬时受体电位M7(transient receptor potential melastain 7, TRPM7)是一种表达广泛、具有通道和激酶特性的双功能膜蛋白,亦被称为通道酶,于2001年首次发现并克隆[1,2] 。近年来的研究表明,脑缺血再灌注损伤过程中,TRPM7通道的过表达和激活导致了细胞内钙超载和ROS的产生,进而促进了神经元的缺氧性死亡;而下调TRPM7表达或抑制其活性能有效逆转上述作用,提示TRPM7与缺血再灌注损伤有密切关系[3]。TRPM7在肾脏组织中表达较丰富,实际上在人类所有器官中肾脏是表达TRPM7较高的器官[4];最新研究表明,TRPM7在大鼠肾脏缺血再灌注损伤中表达上调,TRPM7 mRNA和蛋白在右肾动脉缺血45 min后再灌注24 h的表达量为最高[5]。上述研究提示,与脑缺血再灌注损伤类似,TRPM7也可能对肾脏缺血再灌注损伤的发生过程有重要影响,深入研究TRPM7参与肾脏缺血再灌注损伤的分子机制对发现其作为有效治疗的干预靶点意义重大,因此,本文就TRPM7在肾脏缺血再灌注损伤中的作用及其机制的研究做如下综述。
TRP离子通道是一个蛋白质大家族,其TRP家族成员的各种蛋白有着广泛的结构特征,多样的表达模式,独特的离子选择性,特殊的门控属性,以及多种功能。TRP家族中的M型(TRPM)为与代谢相关的亚家族,而在TRPM亚家族中,目前已知的除TRPM6外,TRPM7是唯一集离子通道属性和激酶属性于一身的"双功能"蛋白。TRPM7的双功能特性在缺血再灌注中作用机制的研究有较大帮助。
TRPM7是由多个亚基组成的同源或异源四聚体,每个亚基由驻留在细胞质内侧的N-末端、C-末端及其间的6个跨膜α-螺旋结构共同组成,见图1(TRPM7通道跨膜结构域组成)。
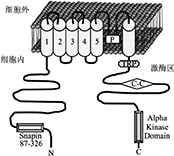
TRPM7每个跨膜结构域由6个相互连接的跨膜α螺旋片段(S1-S6)共同组成,其中S5与S6之间的连接片段呈孔袢(p-loop)结构,胞内区N-末端包含多个蛋白如snapin的结合位点,C-末端包含TRP结合域,C-C(coiled-coiled)区域以及α-激酶区域等重要结构[1]。
TRPM7N-末端结构的改变或者缺失会影响离子通道的活性[1]。TRPM7C-末端包括一个最靠近跨膜结构域称之为TRP框,一个远离跨膜结构区域的非典型α激酶结构域和二者之间的一个卷曲螺旋结构域。C-末端胞内区最重要的一个结构与丝氨酸/苏氨酸蛋白激酶很相似,这一特殊的结构属于α-激酶家族成员,可参与TRPM7的自身或底物磷酸化[1]。
在体内TRPM7各亚基中固定的α-螺旋结构共同围成一个疏水性孔道,可以容许Ca2+、Mg2+、Zn2+、Na+、K+等多种单价或二价阳离子通过,但不能通透阴离子。TRPM7作为一种离子转运膜蛋白,可以选择性地让多种阳离子通过,如信号分子Ca2+、Mg2+、Zn2+,多价阳离子如Gd3+等以及多种微量离子。生理状态下,TRPM7对这些离子的通透性受离子本身及其细胞内外环境的影响而变化。
Arts等[2]首次在神经元氧糖剥夺/复氧(oxygen glucose deprivation/reperfusion, OGD/R)模型上发现了一个奇怪的现象:即在OGD/R晚期联合运用多种离子通道阻断剂也不能抑制细胞内的钙超载,但TRPM7小干扰核苷酸(siRNA)可以有效抑制此钙超载现象,进而阻止神经元的损伤和死亡,该实验表明,TRPM7通道介导的钙超载是OGD晚期引起神经元损伤和死亡的重要因素。此后的一段时间,许多研究者对TRPM7引起的细胞内钙超载现象及其机制做了更加深入地研究发现,TRPM7可通过自身Ca2+通道的开放,使细胞外Ca2+内流,触发内质网三磷酸肌醇(inositol trisphosphate,IP3)受体激活,释放出更多的Ca2+造成细胞内钙超载。除此之外,TRPM7带有一个激酶结构域,也可以直接与磷脂酶(phospholipase C, PLC)结合,水解磷酯酰肌醇二磷酸(phosphatidylinosital biphosphate,PIP2)产生IP3,从而直接调节IP3受体的活性,进而调节细胞内的Ca2+浓度[7]。
体内的Mg2+稳态主要由胃肠道和肾脏来维持。近年来,以TRPM7为代表的膜通道蛋白对Mg2+调节的研究有了较大的进展,将人们对细胞内Mg2+调节机制的认识带到了一个新的高度。Nadler[8]发现TRPM7参与调节和维持细胞内Mg2+平衡和Mg-ATP水平。进一步的研究表明,在PIP2/PLC途径中PLC的激活和PIP2的水解可进一步激活TRPM7通道活性,最终调节Mg2+的跨膜细胞转运[9]。
TRPM7自身的磷酸化可以促进其底物的磷酸化,而且TRPM7的自身磷酸化并不影响其激酶区对底物磷酸化的活性[10]。已有研究证明,TRPM7可通过其上调底物Annexin-1的表达,促进Annexin-1的核、膜转位,加剧OGDR引起的细胞损伤和凋亡。除此之外,TRPM7通过与Annexin-1的相互作用,还可发挥抗炎、免疫应答等作用[11]。同时,TRPM7通过磷酸化底物Calpain和肌球蛋白(Myosin) ⅡA重链(heavy chain,HC)也参与了细胞黏附、细胞骨架的形成、细胞收缩等重要生理过程。研究者检测到激酶区突变的TRPM7电流降低和细胞内ATP的消耗量减少,因此认为TRPM7激酶活性对其离子通道特性是至关重要的。到目前为止,Mg2+被认为是较有可能调节激酶结构域磷酸转移酶活性的物质[12]。
缺血再灌注损伤始于能量代谢障碍及线粒体受损,牵涉多方面的病理改变。其中细胞内Ca2+超载、ROS增加、自身炎性反应机制的激活以及细胞凋亡等机制一直是研究的热点。而目前研究已表明TRPM7涉及以上一系列过程,并在其中发挥着重要作用,下面简要概述一下到目前为止所发现的TRPM7在这一系列过程中的作用及机制。
在缺血再灌注损伤过程中,ROS大量生成,超过了体内酶的清除能力,引起组织广泛性损伤。TRPM7虽然同时表达在肾小管和肾小球,而在缺血再灌注损伤中大多数TRPM7在肾小管被检测到[4]。在急性缺血再灌注损伤组与对照组相比,肾小管上皮细胞钙超载程度及肾细胞凋亡率都显著升高,且与再灌注时间呈正相关。在接下来的一系列研究中又有人发现抑制TRPM7可减轻缺血再灌注损伤[4]。Dusmez等[13]发现Ca2+通道阻滞剂维拉帕米和利多卡因均可以抑制大鼠缺血再灌注损伤所致的TRPM7升高,因缺血再灌注损伤所致的肾小管上皮细胞肿胀,空泡的形成及细胞坏死的情况都有所改善。综上所述,TRPM7在组织缺血再灌注损伤中通过通道和激酶活性,增加细胞内Ca2+浓度,导致组织损伤;而运用TRPM7 siRNA和TRPM7抑制剂如2-APB等,可通过减少细胞内钙超载从而有效地减轻缺血再灌注损伤,也进一步证实了这一点。
在急性肾脏缺血再灌注损伤中肾小管上皮细胞为最易受损伤的部位;然而近十几年来,人们越来越倾向于肾脏缺血再灌注损伤是肾血管系统与小管间复杂的动态相互作用所致,而且血管内皮损伤和肾小管细胞功能障碍还被认为是一系列肾移植缺血再灌注损伤炎症过程的起始点[14]。Meng等[15]发现通过给肾移植小鼠直接注射TRPM7-shRNA,可显著改善肾功能,肾小管损伤和间质纤维化均降低,肾小管坏死、凋亡和炎症的状况也有所改善。在NRK-52E细胞的缺氧模型中,TRPM7-shRNA明显降低活化的P38、MAPKs、Bax和肌动蛋白信号,但是炎症因子与TRPM7表达之间的相互关系尚未有统一的结论。例如Sontia等[16]发现在高醛固酮刺激下小鼠的肾脏组织纤维化及炎症因子IL-6、VCAM-1、COX-2明显增加,而TRPM7及其抗炎底物分子Annexin-1的表达是降低的,故其推测高醛固酮所致的肾损伤是通过TRPM7表达降低,进而引起低镁所致;但Sontia未继续使用TRPM7抑制剂进一步进行验证TRPM7在肾脏炎性损伤中的作用。Yogi[17]通过对大鼠血管平滑肌细胞研究发现:用缓激肽(bradykinin,BK)诱导炎症损伤时,炎性因子表达增加的同时伴随着TRPM7表达的升高,而抑制TRPM7后炎性因子表达有所下调。Sontia与Yogi对TRPM7表达的研究结果不尽一致,这可能与所用诱导剂不同,还有一个是在动物水平,而另一个是在细胞水平,以及所研究器官组织不同所引起。Yogi等[18]随后通过稳转野生型人TRPM7和TRPM7激酶区缺失的质粒进入肾HEK293细胞,对TRPM7的离子通道功能和激酶区功能在炎症中的作用做了进一步研究,结果表明经醛固酮诱导的Mg2+浓度增加及ROS产生增多,两者与TRPM7通道相关,但与激酶区是否缺失并无关联;而在野生型人TRPM7细胞中,加入TRPM7通道抑制剂2-APB后,醛固酮引起的促炎效应被放大;但在激酶区缺失的细胞中,该现象不复存在,故得出结论:不仅TRPM7离子通道与促炎信号通路的抑制相关,TRPM7激酶区也可独立抑制促炎信号通路。
在肾脏缺血再灌注损伤中VCAM-1分子等的表达与肾损伤的严重程度呈正相关[19]。而在炎症过程中,抑制TRPM7不仅可降低IL-6、VCAM-1、COX-2等的表达和减少间质纤维化减轻炎症反应,也可通过其激酶区对促炎信号分子和下游蛋白的抑制或者抑制MAP通路调节抗炎底物Annexin-1进而发挥抗炎作用。有研究表明在冷冻液中加入p38抑制剂后,能减轻心、肺、肝移植中的缺血再灌注损伤[20]。故我们有理由相信对TRPM7的进一步研究有助于开辟新的减轻炎症损伤的途径,尤其是减轻肾脏缺血再灌注损伤。
在缺血再灌注损伤中导致组织和细胞死亡的一个重要原因就是细胞凋亡。细胞内钙离子的超载,凋亡相关分子的相互作用和凋亡相关蛋白水解酶(caspases)的活化均是凋亡的重要过程。江慧[11]等发现:虽然激酶区缺失的TRPM7与表达全长的TRPM7均可显著加重OGD/R引起的HEK293细胞损伤和细胞凋亡,但随着氧糖剥夺后再氧合时间的延长,表达TRPM7全长细胞的损伤程度和细胞凋亡率与激酶区缺失的细胞相比明显加重。这表明TRPM7不仅参与了OGD/R引起的细胞损伤,而且随着再氧合时间的延长TRPM7激酶区在OGD/R引起的细胞凋亡中发挥越来越重要的作用。Ishido曾发现TNFα引起的猪肾LLC-PK1细胞凋亡就是由Annexin-1核转位介导[21]。最新研究表明[22],通过敲除TRPM7基因及病毒转染TRPM7-shRNA等方法,发现TRPM7-shRNA大大减少了Fas受体介导的细胞凋亡以及caspase-3的激活,证实TRPM7参与了Fas受体介导的细胞凋亡。而且TRPM7在细胞凋亡过程中可被caspase在D1510位点裂解为分别包括激酶区域(约40 kD)和离子通道区域(约160 kD)的两部分,裂解后的离子通道活性被大大增强。在通过给予细胞色素C和dATP耗尽caspase-3/8后,这种裂解作用消失,说明TRPM7可加强Fas途径介导的caspase-3的激活,而caspase-3反过来又能促进TRPM7的裂解,增加通道活性,整个过程一起促进了细胞凋亡的发生。而且主要是TRPM7的离子通道功能而非激酶功能在此处发挥了作用。随后,进一步研究表明被裂解的TRPM7的激酶区在胞浆中还可进一步进入细胞核内与核蛋白相结合或者进一步磷酸化组蛋白H3[23]。
2007年人们发现电刺激可通过原肌球蛋白相关激酶A (TrkA)-磷酯酰肌醇3激酶(PI3K)途径抑制缺血再灌注损伤所致的TRPM7表达的升高[24];与此类似,2008年Jiang等[25]发现神经生长因子(NGF)也能通过TrkA-PI3K途径抑制缺血再灌注损伤所致的TRPM7表达的升高。抑制TRPM7后可显著减轻缺血再灌注过程中导致的细胞损伤和凋亡已在多个器官中得到验证。因此,通过TrkA-PI3K途径抑制TRPM7的表达进而抑制凋亡很可能是一种缓解缺血再灌注的细胞损伤的新方法。
TRPM7广泛分布于各种器官组织,在许多刺激下如缺血再灌注损伤时,都有着表达和功能的改变,继而通过参与钙超载和ROS形成、炎症因子的产生和调节、凋亡相关酶以及底物激活等过程,在缺血再灌注损伤甚至是恢复期发挥着重要的作用。随着TRPM7功能被广泛研究的同时,人们近年来越来越多开始探讨TRPM7的作用机制,包括在肾脏缺血再灌注损伤中的机制。无论是通过BK-B2-PLC-Ca2+途径调节TRPM7表达[17]来影响钙超载,还是抑制TRPM7来减轻炎症反应,抑或通过TrkA-PI3K途径抑制TRPM7离子通道和激酶区的功能来干预细胞凋亡的发生,都预示TRPM7将成为减轻缺血再灌注损伤尤其在肾脏的新途径。

























