
研究阻断肝细胞中RIPK3(receptor-interacting protein kinase 3)诱导的坏死性凋亡对小鼠肝脏缺血再灌注损伤(IRI)的影响。
60只小鼠采用Stata统计软件随机分为4组:野生型(WT)对照组、肝特异性敲除RIPK3(HKO)对照组、WT-IRI组,HKO-IRI组。对照组进行假手术,即开腹后仅游离肝门部血管,不阻断血流,WT-IRI组和HKO-IRI组开腹后游离肝门血管并阻断肝左叶和肝中叶的血供90 min,然后开放血管6 h,取各组小鼠的血液和肝组织样本,检测肝功能,免疫组织化学及HE染色检测炎症浸润及肝损伤,蛋白质印迹法检测自噬相关蛋白LC3-Ⅱ、P62。并提取WT小鼠和HKO小鼠原代肝细胞,分为对照组和缺氧复氧组(HIR组)。贴壁后HIR组缺氧4 h,复氧4 h,取上清液检测ALT、AST,细胞提取蛋白检测LC3-Ⅱ、P62。
与对照组相比,IRI组小鼠肝组织明显受损,但是与WT-IRI组相比,HKO-IRI组肝损伤显著加重(P<0.05),进一步研究发现其肝组织中LC3-Ⅱ蛋白含量明显下降,P62蛋白含量增加。同样,肝细胞缺氧复氧后,相对于WT鼠,HKO来源的肝细胞损伤也更重。
RIPK3敲除阻断肝细胞坏死性凋亡后,通过抑制自噬加重了肝IRI。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
缺血再灌注损伤(ischemia reperfusion injury,IRI)广泛存在于临床手术中,是影响肝移植受者长期存活的重要因素。肝细胞的死亡是其中的重要环节,而坏死性凋亡这种死亡方式是否在其中发挥重要作用尚不清楚。之前的研究表明坏死性凋亡在肝IRI中起重要作用[1,2],RIPK3是引起坏死性凋亡的重要分子[3,4]。因此我们通过肝特异性敲除RIPK3抑制肝细胞坏死性凋亡观察其在肝脏IRI中的作用。
雄性WTC57BL/6小鼠(6~8周龄),无固定病原体级,购自上海斯莱克实验动物有限公司。肝特异性敲除RIPK3(HKO)小鼠(6~8周龄)购自上海南方模式生物科技股份有限公司。
ALT/AST检测试剂盒、TNF-α和IL-6 ELISA试剂盒购自南京建成生物工程学院,LC3-Ⅱ、P62 、MPO抗体购自美国Cell Signaling公司,F4/80购自美国AbD Serotec公司。
取WT鼠30只采用Stata统计软件随机分为2组,野生型(WT)对照组和WT-IRI组,每组15只,同样取HKO鼠30只分为2组,HKO对照组和HKO-IRI组,每组15只。我们采取热缺血再灌注模型[5],对照组开腹后仅游离肝门部血管,不阻断血流,WT-IRI组和HKO-IRI组开腹后游离肝门血管并阻断肝左叶和肝中叶的血供90 min,然后开放血管6 h。所有手术均有同一术者完成,小鼠术前禁食14 h。
按照文献报道的方法进行原代肝细胞分离[6],即用1%的戊巴比妥钠腹腔注射麻醉小鼠后,固定小鼠,打开腹腔,暴露出肝脏及门静脉,用30 ml 37 ℃预温的1 mmol/L EGTA进行原位肝灌注,直至肝内血液流尽。再用37 ℃预温的0.75 g/L的I型胶原酶溶液原位灌注,直至肝脏变软,呈乳黄色。然后切下肝脏放入盛有0.08 g/L I型胶原酶的60 mm培养皿中,将肝组织钝性分离至糊状,用滤网(70 μm)过滤至无菌50 ml离心管中,4 ℃,50 ×g离心5 min,弃上清,加入30 ml GBSS重悬后再次离心,沉淀加入培养基重悬,细胞计数,将分离的细胞接种在培养皿6孔板中(2×105细胞/孔),3 h后换液。
将铺好的肝细胞在37 ℃低氧培养箱中(内含5%CO2,90%N2和5%O2)培养4 h,然后将肝细胞放回到含氧量正常的37 ℃培养箱中4 h。
术后6 h采集各组受鼠腹主动脉血,离心获取血清。收集肝细胞对照组和缺氧复氧组的上清液。使用ALT/AST检测试剂盒检测血清细胞上清液的丙氨酸转氨酶(ALT)和天冬氨酸转氨酶(AST)水平。
采用蛋白质印迹法检测。术后6 h,取各组肝组织200 mg,加入蛋白质裂解液和蛋白抑制剂苯甲基磺酰氟化物,于4 ℃下匀浆,12 000 ×g离心30 min,取上清液中蛋白质,定量后变性,行聚丙酰胺凝胶电泳,转移至硝酸纤维膜,室温封闭1 h,于4 ℃下过夜,洗涤后加入兔抗LC3-Ⅱ或P62多克隆抗体(1∶1 000),于4℃下过夜,洗涤后加入辣根过氧化酶标记的抗兔IgO(1∶5 000),用化学发光试剂检测,并用X线片显影。
将肝组织在40 g/L多聚甲醛中固定至少24 h,然后通过标准程序石蜡包埋并切成5 μm厚的切片。对于肝脏组织病理学,切片用HE染色。Suzuki的标准用于评估肝损伤的组织学严重程度。对于IHC分析,首先将肝切片再水化并加工用于抗原暴露,然后与抗MPO或F4/80的一抗中4℃孵育过夜,然后孵育辣根过氧化物酶偶联的二抗。结果由病理学家评估切片。
使用ELISA试剂盒检测小鼠血清的IL-6、TNF-α。
应用SPSS软件16.0进行统计学处理,实验结果以Mean±SD表示,多组间均数的差异性比较用单因素方差分析,两两比较用t检验,P<0.05为差异有统计学意义。
与假手术组相比,IRI组的血清ALT和AST水平均明显升高(P<0.05,表1),与WT-IRI组相比,HKO-IRI组血清ALT和AST水平升高更为明显(P<0.05,表1)。
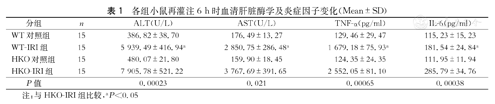
各组小鼠再灌注6 h时血清肝脏酶学及炎症因子变化(Mean±SD)
各组小鼠再灌注6 h时血清肝脏酶学及炎症因子变化(Mean±SD)
| 分组 | n | ALT(U/L) | AST(U/L) | TNF-α(pg/ml) | IL-6(pg/ml) |
|---|---|---|---|---|---|
| WT对照组 | 15 | 386.82±38.70 | 176.49±13.27 | 129.46±29.47 | 115.23±15.23 |
| WT-IRI组 | 15 | 5 939.49±416.94a | 2 850.75±286.48 a | 1 679.18±75.93 a | 181.54±24.84a |
| HKO对照组 | 15 | 480.07±21.80 | 159.90±18.45 | 124.35±24.35 | 111.95±11.94 |
| HKO-IRI组 | 15 | 7 905.78±521.22 | 3 767.69±391.65 | 2 552.05±81.10 | 285.79±34.76 |
| P值 | 0.00023 | 0.021 | 0.00065 | 0.00038 |
注:与HKO-IRI组比较,aP<0.05
与假手术组相比,IRI组的血清TNF-α和IL-6水平均明显升高(P<0.05,表1),与WT-IRI组相比,HKO-IRI组血清TNF-α和IL-6水平升高更为明显,差异具有统计学意义(P<0.05,表1)。
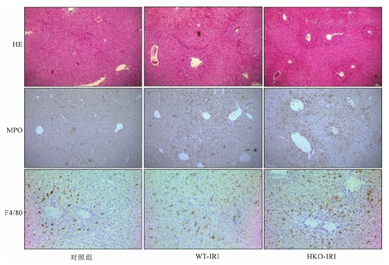
HE染色发现与对照组相比,IRI组出现肝损伤区域,并且HKO-IRI组损伤区域明显大于WT-IRI组(P<0.05,图1),IHC染色表明与对照组相比,IRI组肝组织中巨噬细胞和中性粒细胞浸润增加,且与WT-IRI相比,HKO-IRI组增加更为明显(P<0.05,图1)。以上结果表明,RIPK3的敲除明显增加了小鼠肝缺血再灌注的损伤。
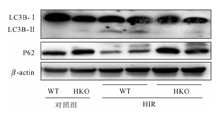
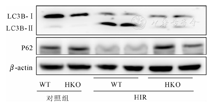
与WT-IRI组相比,HKO-IRI组小鼠肝脏组织中的LC3-Ⅱ蛋白表达量明显减少而P62蛋白表达量明显增加(图2),这表明相比于WT-IRI组,HKO-IRI组的自噬明显减轻。即敲除RIPK3抑制了自噬,从而加重了肝脏IRI。
提取WT小鼠和RIPK3敲除小鼠的原代肝细胞,贴壁后做缺氧复氧实验,相比于WT鼠的原代肝细胞,HKO鼠的原代肝细胞缺氧复氧损伤更为严重严重,上清中ALT/AST的含量更高(P<0.05,表2)。与WT-HIR组相比,HKO-HIR组小鼠肝细胞中的LC3-Ⅱ蛋白表达受到抑制,而P62蛋白含量明显增加(图3),表明缺氧复氧后,相对于WT-HIR组,HKO-HIR组小鼠肝细胞自噬,明显减少。
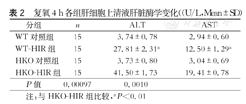
复氧4 h各组肝细胞上清液肝脏酶学变化((U/L,Mean±SD)
复氧4 h各组肝细胞上清液肝脏酶学变化((U/L,Mean±SD)
| 分组 | n | ALT | AST |
|---|---|---|---|
| WT对照组 | 15 | 3.74±0.78 | 2.94±0.60 |
| WT-HIR组 | 15 | 27.81±2.31a | 12.50±1.29a |
| HKO对照组 | 15 | 3.73±0.80 | 3.04±0.69 |
| HKO-HIR组 | 15 | 41.50±1.73 | 19.41±0.78 |
| P值 | 0.00097 | 0.0010 |
注:与HKO-HIR组比较,aP<0.01
肝IRI一直是临床上影响肝脏外科手术预后的重要问题,其限制了肝切除及肝移植的临床应用。肝IRI是由多种细胞和因子介导的复杂过程,凋亡、坏死、坏死性凋亡、自噬等多种死亡方式参与其中,并且相互影响[7,8]。因此对IRI机制和新治疗靶点的进一步研究显得至关重要。
有研究表明,坏死性凋亡在肝IRI中起重要作用[9]。RIPK3是引起细胞坏死性凋亡的重要因子[5]。磷酸化的RIPK3与RIP1结合形成诱导坏死性凋亡的复合体,复合体激活下游的混合系列蛋白激酶样结构域(MLKL),激活的MLKL作用于细胞膜使细胞裂解坏死并释放炎症因子[10]。本研究发现,在小鼠肝IRI模型中,RIPK3肝特异性敲除小鼠的血清ALT、AST水平较对照小鼠明显升高,炎症因子TNF-α、IL-6也有所增加,表明RIPK3敲除后,使小鼠肝脏IRI加重,RIPK3是肝IRI机制中的关键因子。
最近的研究表明自噬和坏死性凋亡密切相关[11]。当用TNF-α激活成纤维细胞系半胱天冬酶-6时,自噬在其中起负调节坏死的作用[12],并且抑制自噬时能减轻棕榈酸诱导的坏死性凋亡[13]。自噬是一种复杂且高度调控的过程,它将细胞物质传递给溶酶体,使其降解、循环和生成促进细胞代谢的分子。自噬被认为是在不同情况下调节细胞损伤的适应性反应[14]。然而,过度的自噬反应直接导致细胞损伤,例如在IRI期间过度自噬介导的心肌损伤[15]。一般情况下,自噬通过与其他细胞死亡信号传导机制相互作用来调节细胞功能。自噬有多种的标记物,目前应用最广泛的是LC3-II[16]。发生自噬时,LC3-I被剪切形成LC3-II,而LC3-II只存在于自噬体内容物中,随着自噬体膜的增加而增加[17],因此,LC3-II可以作为自噬的检测指标。在本课题研究中,我们检测了LC3B-II的水平,发现在肝脏IRI之后LC3B-II的水平显著增加,但是相较于RIPK3敲除鼠,野生型小鼠肝脏LC3B-II水平增加更明显,表明在肝IRI中,敲除鼠的自噬相对较轻。同时P62的蛋白表达水平增加,也表明HKO-IRI组的自噬减轻。
本研究发现,HKO-IRI组的ALT/AST、TNF-α、IL-6血清含量增加比WT-IRI组更为明显,说明自噬抑制加重了肝缺血再灌注肝损伤。髓过氧化物酶(MPO)是中性粒细胞特异性酶,而F4/80是巨噬细胞的特异性酶,它们的活性是反映中性粒细胞和巨噬细胞浸润的重要指标之一,我们通过IHC染色发现敲除RIPK3诱导的自噬抑制也加重了炎症细胞的浸润,这在肝IRI中起重要作用。我们的体外实验进一步表明敲除RIPK3后抑制肝细胞的自噬,加重肝细胞缺氧再复氧损伤。
综上所述,本实验研究表明敲除RIPK3抑制坏死性凋亡后会抑制肝细胞自噬,增加炎症细胞浸润,从而加重肝脏IRI。本研究探究了RIPK3在肝脏缺血再灌注中的作用及RIPK3诱导的坏死性凋亡和自噬的关系,为今后进一步研究RIPK3在肝脏IRI的分子机制及临床应用提供了理论基础,对临床上预防肝脏IRI有重要意义。
所有作者均声明不存在利益冲突

























