
本文就门静脉性肺动脉高压(portopulmonary hypertension,PoPH)发病机制、治疗、管理等多个方面的最新研究进行综述,以期为临床肝移植术前评估和治疗提供参考。PoPH患者死亡原因以肝脏原发疾病为主,肝移植是主要的治疗手段,术前针对肺动脉压力的评估及治疗可能是影响肝移植预后的主要因素。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
门静脉性肺动脉高压(portopulmonary hypertension,PoPH)是由肝内病因(肝硬化、肝结节等)或肝外病因(肝外门静脉阻塞、胆管闭锁等)引起的门静脉高压进而出现肺动脉压力增高。PoPH是门静脉高压的罕见并发症。研究表明,1年、3年和5年的存活率分别为77%、52%和34%[1]。并且PoPH患者住院率及花费更高[2]。Bosch等[3]研究表明肝静脉压力梯度(HVPG)≥10 mmHg考虑门静脉高压症(portal hypertension,PHT),并可以预测食管胃底静脉曲张的发展。
近年来,随着临床诊断手段的提高和肝移植手术的不断成熟,PoPH的诊断率提高,PoPH患者行肝移植手术也可以获得较好的结果。但目前PoPH的发病机制及治疗等方面尚不明确,现就PoPH发病机制、治疗、管理等多个方面的最新研究进行综述。
肝硬化和门静脉高压导致内脏血管扩张和门体分流形成,有多种方式促成PoPH发病机制[4]。主要分为以下几个方面。
肝纤维化导致门静脉高压、肝血流阻力增加和内脏血管舒张。内脏血管舒张导致人体上半部分的循环量增加。并且通过门体分流将血流从肝脏转移至心脏,导致整体高动力循环状态。如肝纤维化未得到及时改善,肝内血液流动的抵抗力增加导致门静脉压力梯度增加,肝血流量减少,通过产生新血管来增加门体侧支,门体分流增加[5]。进而导致食管胃底静脉曲张、腹壁静脉曲张、痣静脉曲张、Retzius静脉曲张、脾肾分流等,随着病情进展会产生腹腔积液、胸腔积液、水钠潴留等,形成恶性循环。一方面大量腹水会使膈肌抬高,伴或不伴有胸腔积液时,肺部受压,活动度减低,导致肺内缺氧,肺小动脉痉挛,肺血管阻力增加,右心室后负荷增加,右心需要更大的射血能力来维持平衡,久而久之导致右心衰竭;另一方面,由于水钠潴留,液体负荷重,心脏前负荷增加。综合这两个方面,根据Frank-Starling机制,均会导致心输出量增加,进而肺血管容量增加,平均肺动脉压增加,进而导致肺动脉高压。(图1)
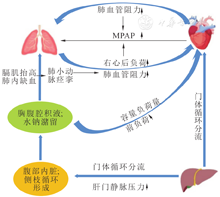
有诸多研究表明,某些在肺循环中起作用的血管活性物质,如肺血管扩张剂吸入型一氧化氮(Inhaled nitric oxide,iNO)和前列环素,以及肺血管收缩剂内皮素-1、血栓素A2和5-羟色胺等,在肝硬化患者中处于失衡的状态[6,7,8]。肺脉管系统中这些血管活性物质的失衡导致血管收缩和肺血管阻力升高。同时,肺血管内皮和下层平滑肌的损伤导致永久性血管重塑,综合起来最终导致肺动脉高压的发展。Humbert等[9]的研究表明,动脉平滑肌的暴露和损伤,将导致平滑肌增殖,以及肺血管内外膜、中间肌层和外膜增厚,而动脉壁增厚可导致肺血流迟缓,血小板聚集和血栓形成。Stenmark等[10]研究认为整个肺动脉血管系统的重塑过程,可能均与动脉血管平滑肌细胞的增生和肥大有关,在重塑过程中可能表现出不同程度的增殖、炎症反应以及细胞外基质的变化。Yang等[11]通过大鼠试验表明,血栓烷A2释放介导甲氧胺刺激门静脉灌注压增加。Türker等[12]通过回顾性分析发现,促甲状腺激素(thyroid-stimulatinghormone,TSH)升高也是PoPH发展的独立危险因素。
Baeyens等[13]的研究表明,在肺动脉内持续高血流量产生的剪切力,导致内皮细胞损伤,以及参与血管重塑过程的基因的激活和抑制,是肺动脉高压形成的有关因素。Bajolle等[14]对1例肺动脉高压患儿的基因研究发现,其肺动脉高压和骨形态蛋白受体2(BMPR2)基因突变有关。并且对肺动脉高压(Pulmonary artery hypertension,PAH)的病例分析发现BMPR2基因突变占15%~40%,且BMPR2基因在家族性肺动脉高压中起关键作用[15,16]。骨形态蛋白9(BMP9)是由肝星状细胞产生的循环因子,是BMPR2信号传导的配体,已被证实对肝纤维化具有保护作用[17,18]。Jiang等[19]通过大鼠实验研究发现,BMP9、BMPR2及SMAD信号通路的抑制,导致肺动脉内皮严重丧失,进而肺动脉血管重塑,导致PH的发展。多项对照研究表明,PoPH的患者BMP9水平降低[20,21]。Liu等[22]通过小鼠实验研究发现rs1042636、rs6776158、rs1048213和rs9883099的次要等位基因的点突变促进了PH的发展和严重程度。总而言之,PoPH可能与BMP9、BMPR2等一些其他基因突变有关。
PoPH的诊断包括对肺循环和肝循环这两方面的血流动力学评估。食管静脉曲张和皮肤侧支静脉曲张提示门静脉高压症。在超声检查中,门静脉血流速度降低和门静脉双相或血流逆转提示门静脉高压。肝静脉压力梯度(hepatic venous pressure gradient,HVPG)即楔形肝静脉压力-游离肝静脉压力>5 mmHg可诊断为窦性门静脉高压。Dajti等[25]通过一项回顾性分析研究表明脾硬度测量结合肝硬度测量值、肝静脉压力梯度可以提高门静脉高压的诊断。在2018年举行的第六届世界肺动脉高压研讨会上,修订了《毛细血管前肺动脉高压诊断的血流动力学标准》。肺动脉高压(pulmonary artery hypertension,PAH)诊断标准为平均肺动脉压(mean pulmonary artery pressure,mPAP)>20 mmHg、肺血管阻力(pulmonary vascular resistance,PVR)≥3 Wood units(WU)(240 dyne·s·cm-5)和肺动脉楔压(pulmonary artery wedge pressure,PAWP)<15 mmHg[26]。
Kawaguchi等[27]通过对833例慢性肝病患者研究发现,mPAP与白蛋白-胆红素(ALBI)评分有显著相关性,并且在ALBI评分≥-1.45的患者中,均表现出严重的PoPH,而在ALBI评分<-1.45的患者中,PoPH的患病率为25.0%。因此,ALBI评分可能有助于评估肺血管阻力。ALBI计算公式为ALBI(数值)=0.66×lg胆红素(μmol/L)-0.085×白蛋白(g/L)[28]。
吸氧可以降低PoPH患者的PVR,《门静脉性肺动脉高压肝移植围手术期管理中国专家共识(2021版)》推荐当PoPH受者外周血氧饱和度<91%或动脉血氧分压<60 mmHg时需要吸氧,使血氧饱和度>92%[29]。
(1)内皮素受体拮抗剂治疗
马西替坦:是一种双重内皮素受体拮抗剂(ERA),本身是来源于波生坦的结构,通过用磺酰胺部分替换波生坦中存在的磺酰胺,从而增加了受体亲和力和亲脂性。建议用量为10 mg/d(口服1次)。与波生坦相比,肝毒性明显降低,并且水肿的频率低于波生坦和安立生坦。但关于对儿童和长期服药的后果仍然存在一些不确定性[30]。Pulido等[31]通过一项随机双盲多中心安慰剂对照实验证实,服用马西替坦组与安慰剂组相比,服用3 mg/d和10 mg/d的马西替坦可有效延缓疾病进展,并且在治疗期间可以将发病或死亡事件的风险降低45%(10 mg/d)和30%(3 mg/d)。
波生坦:是非选择性内皮素受体拮抗剂。Savale等[32]回顾性研究证实波生坦可以改善Child-Pugh B级肝硬化患者的血流动力学。但由于波生坦有肝功能损害的副作用,对于严重肝功能不良者应慎用。Muraoka等[33]通过对PoPH患者应用低剂量波生坦联合低剂量安立生坦,可以有效降低门静脉性肺动脉高压,并且还可以最大限度的减轻波生坦的肝功能损害和安立生坦导致外周水肿的副作用。Usta等[34]通过自纳米乳化药物传递系统,可以优化波生坦由于水溶性低而导致的口服生物利用度差的问题。Köse等[35]通过动物实验表明,波生坦不仅有治疗肺动脉高压的作用,同时也能预防骨质疏松症的发展。
安立生坦:是一种选择性内皮素受体A拮抗剂。Cartin-Ceba等[36]通过前瞻性研究证实,在中重度PoPH患者中,安立生坦可以显著改善肺血流动力学反应,并且对肝功能损害小。Preston等[37]对初次诊断的Child-Pugh A/B级的PoPH患者,给予安立生坦24周及用药期延长(24~28周)治疗后,发现安立生坦单药治疗PoPH可以在用药24周时改善血流动力学。但用药期间有38.7%发生水肿,22.5%发生头痛,其中有1例下肢水肿导致永久停药。所以针对这些内皮素受体拮抗剂的不良反应,可以考虑波生坦和安立生坦低剂量联合治疗[33],在改善肺血流动力学的同时,减轻不良反应。
(2)PGI2及其类似物
前列环素及其类似物主要由血管内皮细胞产生,对血管具有较强的扩张作用,可以选择性作用于肺循环,降低肺动脉压力和肺血管阻力,为降低肺动脉压的首选药物。主要代表药物有前列腺素E1、曲前列环素、贝前列环素以及吸入用伊洛前列素。近期对PGI2的研究较少,最近的有2012年Touma等[38]报告证实依前列醇在治疗PoPH时可导致脾功能亢进,但停药后可逆转。对有PoPH患者拟行肝移植术前早期静脉应用前列环素类似物,来降低肺血管阻力,改善右心功能,有助于肝移植的顺利进行[39,40]。
(3)5型磷酸二酯酶抑制剂
5型磷酸二酯酶抑制剂可以特异性地抑制磷酸二酯酶,使环磷酸鸟苷(cGMP)降解减少,cGMP激活cGMP激酶,钾通道开放,引起血管舒张。常用的药物有西地那非、他达拉非等。Rosario等[41]研究表明,西地那非可以对PoPH患者的右心收缩和舒张功能有积极影响,并且能够调节右心的反向重塑功能。Cheng等[42]报道3例肝移植术前诊断为重度PoPH,术前经过西地那非单药治疗使平均肺动脉压力降低34%~47%,未出现相关的不良反应,并且成功行肝移植手术。有报道肝移植术后伴有肝肺综合征和门静脉高压时,应用西地那非也是有效且安全的[43]。
(4)其他相关药物
除了靶向PAH的药物治疗外,还需要同时进行其他支持性治疗。包括利尿剂、β受体阻滞剂、血管紧张素受体阻滞剂,伴有右心功能不全时还可合用洋地黄类药物[44]。对于合并右心功能不全、中心静脉压升高、四肢水肿、腹水、肝淤血等相关临床表现的PoPH患者,可使用利尿剂缓解水钠潴留症状,但结合患者血压、电解质等临床表现适当应用[29,45]。根据患者情况,可加用普通肝素或低分子肝素预防肺微小动脉原位血栓形成。但PoPH患者常伴有肝功能不全、脾功能亢进,凝血功能差,目前尚无证据支持抗凝治疗可使PoPH患者获益,所以《2021版门静脉性肺动脉高压肝移植围手术期管理中国专家共识》不建议常规抗凝治疗[29]。Breen等[46]研究表明,磺酰脲受体亚基1(SUR1)通过激活可以调节肺血管张力,减缓肺动脉高压的进展,所以SUR1激活剂可以作为治疗PoPH的一个研究点。非选择性β受体阻滞剂是治疗肝硬化门静脉高压的基本用药,非选择性β受体阻滞剂和内窥镜套扎术用于中、重度静脉曲张出血的初级预防[47]。虽然非选择性β受体阻滞剂可以降低门静脉压力预防消化道出血,但是停用β受体阻滞剂后mPAP≥35 mmHg的PoPH患者6 min步行距离增加及血流动力学好转,因此不推荐中重度PoPH患者使用β受体阻滞剂[29,48]。
PoPH患者行经颈静脉肝内门体分流术(transjugular intrahepatic portosystemicshunt,TIPS)治疗前,需严格评估心脏功能。因行TIPS术后PoPH患者的右心前负荷、心输出量增加,致肺血管压力增加、血管阻力升高,发生右心衰竭风险增加。《2016年国际肝移植学会实践指南》以及《2021年门静脉性肺动脉高压肝移植围手术期管理中国专家共识》均指出充血性心力衰竭和重度肺动脉高压(mPAP≥45 mmHg)是TIPS的绝对禁忌证;中度PoPH(35 mmHg≤mPAP<45 mmHg)是相对禁忌证[29,49]。
(1)肝移植的风险与选择
肝移植术是肝功能衰竭患者的最终选择,但存在肺动脉高压的患者行肝移植手术时风险更高。早在2000年,Krowka等[50]将PoPH患者根据mPAP严重程度分为轻、中、重三组。在这三组中,重度PAH组行肝移植术死亡率为100%,中度组死亡率为50%,轻度组无死亡报告。所以较高的mPAP,对肝移植来说有较高的死亡风险。严重肺动脉高压是肝移植的禁忌证,如需行肝移植术,术前需改善肺脏血流及改善右心功能。Lee等[51]通过对1 307例肝移植的肝硬化受者回顾性分析,发现严重PH的受者1年的移植存活率为87%,指出术中评价肺动脉压力有助于预测肝移植后的早期临床结局。
美国肝病研究学会推荐的考虑进行肝移植的肝病患者的一般原则:所有接受肝移植评估的患者均需通过经胸超声心动图筛查肺动脉压力,如果右心室收缩压>45 mmHg的患者,应通过右心导管插入术进一步评估[52]。mPAP<35 mmHg的患者不需要PAH特异性治疗,可以进一步评估是否可以行肝移植手术。mPAP>35 mmHg的患者应由经验丰富的相关专家进行评估,并考虑进行PAH特异性治疗。如果mPAP可以降低到<35 mmHg并且PVR可以通过药物治疗降低到<5 WU,则可以进行肝移植(图2)。门静脉性肺动脉高压者接受肝移植时心血管和总体死亡风险增加,但终末期肝病模型(model for end-stage liver disease,MELD)实验室评分中并未纳入这种风险,同时对于有轻度肝病和PoPH的患者,后期死亡率较高的风险也未被纳入到MELD评分中[53]。所以2021年美国器官采购和移植网络批准,满足以下标准患有PoPH的肝移植等待者有资格获得MELD加分:初始mPAP>35 mmHg和PVR>3 WU;患者接受PAH特异性治疗;并且在治疗后mPAP降至<35 mmHg[54]。
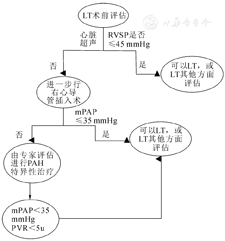
注:LT为肝移植;RVSP为右心室收缩压;PAH为肺动脉高压;mPAP为平均肺动脉压;PVR为肺血管阻力
(2)肝移植的获益
Savale等[53]的研究回顾分析35例PoPH肝移植受者的血流动力学和生存数据,发现肝移植后6个月、1年和3年的存活率分别为80%、77%和77%,其中存活超过6个月的27例受者全部停用静脉注射依前列醇,大多数受者在肝移植术后的血流动力血有所改善,30%的受者在最后一次随访时mPAP<25 mmHg。该数据表明,PoPH受者经过肝移植治疗后,门静脉性高压解除,门体分流逐渐减少,血流动力学改善,肺动脉压力逐渐下降。
PoPH是一个具有挑战性的疾病,5年存活率较低,与潜在肝病的严重程度密切相关,而与PoPH的血流动力学严重程度无关[1]。Sahay等[55]对69例PoPH患者死亡原因分析,其中13例由PoPH直接或间接导致的死亡。以上多项研究表明,PoPH患者的主要死亡原因以肝脏原发病为主。PoPH有如下方面需要进一步研究:如何早期诊断PoPH,并且做出严重程度分型,并提供个体化治疗方案;在PAH降至何水平后评估肝移植手术可以带来更高的成功率;目前国际上还没有关于PoPH特异性治疗PAH或是否在肝移植后继续治疗PoPH的指南推荐。
门静脉性肺动脉高压是门静脉高压的罕见并发症,发病机制有多方面,包括形成高动力循环状态、血管活性物质的相互作用、BMP9及BMPR2等的相关基因突变、雌激素的作用等。但有很多研究只是在动物实验阶段,对人类的影响还需要进一步探究。然而在预测评分方面,ALBI评分可能有助于评估肺血管阻力。内皮素受体拮抗剂、PGI2及其类似物、5型磷酸二酯酶抑制剂等在治疗PoPH方面仍发挥重要作用,PoPH患者的主要死因是肝脏原发病,肝移植是主要的治疗手段,术前评估肺动脉压力以及术前的干预治疗,对肝移植手术可以获益。
所有作者均申明不存在利益冲突

























