
版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
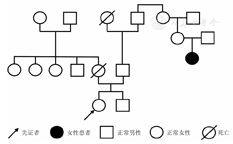
患者,女,25岁,主因发现血小板减少2年,月经增多3个月,牙龈出血10 d,为进一步治疗入住本院。体检:体温36.6 ℃,脉搏80次/min,呼吸20次/min,血压115/82 mmHg(1 mmHg=0.133 kPa)。全身皮肤黏膜无黄染、无出血点,少量陈旧性瘀斑;浅表淋巴结未触及;咽无充血,扁桃体不大;胸骨无压痛,双肺呼吸音清,未闻及干、湿罗音,无胸膜摩擦音。肺CT、头颅CT未见异常。心率80次/min,律齐,心音有力,各瓣膜听诊区未闻及杂音,无心包摩擦音。腹平软,肝脾肋下未触及。既往5年前患胃溃疡(胃镜证实),否认2型糖尿病、高血压病史,无肾脏损害、听力障碍及白内障。患者有一位亲属发现血小板减低。实验室检查:血常规:Hb 147 g/L,Ret 60.7×109/L,WBC 5.49×109/L,PLT 27×109/L。外周血涂片未见幼稚细胞,部分成熟粒细胞胞质内可见大小不一的蓝斑,巨大血小板易见(图1)。骨髓象:有核细胞增生活跃。粒系各期均见,以中晚幼细胞为主,部分成熟粒细胞胞质内可见一个或多个圆形或不规则形淡蓝色包涵体,可见巨大血小板(图2)。电子显微镜检查:结果可见粒细胞内包涵体呈横行分布的核蛋白体,并有纵向微丝相连(图3)。免疫分型:流式细胞术检查以CD45细胞侧向角散射光强度(SSC)设门,CD3+细胞占淋巴细胞比例:76.5%,CD3+CD4+占淋巴细胞比例:33.0%,CD3+CD8+占淋巴细胞比例:40.3%,CD3+CD4+/CD3+CD8+:0.82。血小板聚集率:ADP诱导率:65.9%,胶原诱导率:66.7%。血块收缩试验:24 h不收缩。凝血常规及肾功能检查均正常。基因测序:应用美国ABI公司的3730XL测序仪对先证者非肌性肌球蛋白重链9(nonmuscle myosin heavy chain 9,MYH9)基因1、10、16、25、26、30、31、38、40号外显子PCR扩增产物进行测序[1,2,3,4],确认突变位点在22号染色体36691607位置(MYH9基因26号外显子的3429处),命名为c.3429 T>G,该突变使相应密码子由GCU变为GCG,命名为p.A1143A,为同义突变(图4)。家系调查显示患者有一位亲属患有血小板减少症,其余无异常(图5)。
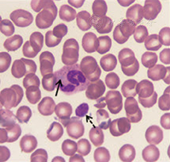
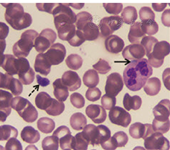
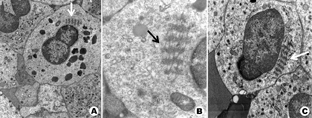
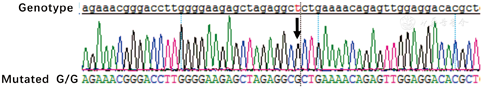
根据临床及实验室检查,诊断为May-Hegglin异常(MHA)。入院后完善相关检查,患者血小板明显降低,不排除合并特发性血小板减少性紫癜(Idiopathic Thrombocytopenic Purpura,ITP),应用激素后效果不明显,停激素治疗。患者一般情况可,要求院外治疗,签字后离院。
MHA在1909年由May首次发现,1945年由Hegglin全面描述。据文献报道,MYH9基因突变会导致常染色体显性遗传病的发生,属于非肌球蛋白重链9相关疾病(nonmuscle myosin heavy chain 9 related diseases,MYH9-RD),临床上除表现为血小板减少、巨大血小板和粒细胞包涵体三联征外,还伴有耳聋、肾炎和白内障,根据临床表现的不同主要分为MHA、Sebastian、Fechtner和Epstein综合征[5]。4种MYH9-RD在发病过程中具有一定的相关性,近来研究发现[6],起初诊断为MHA和Sebastian综合征的仅有血液系统表现的患者,可逐步表现出Fechtner综合征和Epstein综合征的肾炎及耳聋,同一家系的患者可以表现为Epstein综合征,也可表现为Fechtner综合征,而且通过对大量已报道的病例评估发现,仅有MHA患者表现出血栓形成的症状。4种MYH9-RD的鉴别见表1。

4种MYH9-RD的鉴别[6]
4种MYH9-RD的鉴别[6]
| MYH9-RD类型 | 血栓形成 | 血小板减少 | 巨大血小板 | 粒细胞包涵体 | 耳聋 | 肾炎 | 白内障 |
|---|---|---|---|---|---|---|---|
| May-Hegglin异常 | + | + | + | + | - | - | - |
| Fechtner综合征 | - | + | + | + | + | + | + |
| Sebastian综合征 | - | + | + | + | - | - | - |
| Epstein综合征 | - | + | + | - | + | + | - |
注:MYH9-RD:非肌球蛋白重链9相关疾病
该病在临床上易被误诊为ITP或巨大血小板病(Bernard-Soulier综合征)。本先证者起病时诊断为ITP,应用激素后效果欠佳,经细胞形态学观察到粒细胞胞质内淡蓝色包涵体及巨大血小板,又经家族史调查发现家族成员有血小板减少症,遗传方式符合常染色体显性遗传(图5),电镜超微结构检查也进一步证实了粒细胞包涵体的存在,该患者同时伴有血小板减少,无耳聋、肾炎和白内障病史,符合MHA诊断。
MHA主要为MYH9基因突变所致,因此该病的诊断除依赖于实验室检测和临床表现外,还需基因测序分析最终确诊。迄今为止,报道的200多个家系已经鉴定出43种不同的基因突变类型,大部分突变为错义突变,且主要集中在1、6、16、26、30、38和40号外显子,覆盖了接近临床85%的病例[7]。国外研究发现E1841K、D1424N或D1424H和R1933X为常见突变类型[1],而国内目前共发现5种突变类型:Q3H、E1841K、V1516L、R1933X和D1424N,其中E1841K为我国常见的突变热点[4]。除错义突变外,个别报道有无义突变、移码突变和缺失突变,但都集中在40号外显子[8,9,10]。徐敏等[11]于2001年报道了我国第一个MHA家系并进行了基因鉴定,发现该家系致病基因突变类型为E1841K。本研究证实先证者MYH9基因的26号外显子存在p.A1143A型同义突变,在国内外尚属首次。由于未引起编码氨基酸的改变,可能直接从基因水平通过影响细胞信号传导等途径使患者出现相应临床症状,但由于此种突变类型报道尚少,其致病机制还需进一步的探究。
MHA的诊断在于临床血液学工作者提高警惕,初步的形态学筛查是减少漏诊和误诊的重要手段,基因诊断方法的建立和应用将有效而特异地确诊MHA,极大地提高临床诊断水平。但MHA目前尚无特效治疗,更深入的分子生物学研究及基因敲除动物模型的建立有利于进一步明确发病机制,开展治疗研究。

























