
本例患儿表现为小头畸形、生长及智能等发育迟缓以及肾病综合征。全外显子测序发现患儿在20号常染色体的45317947和45315426位置上分别发生c.107T>C和c.728G>T突变。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
Galloway-Mowat综合征(GAMOS)是一种肾脏、神经系统受累的常染色体隐性遗传(AR)病,该病临床主要表现为小头畸形、发育迟缓及肾病综合征等。患儿多在母体宫内即出现发育迟缓且伴随有早产现象。虽然目前国内外均已有对该病的临床病例诊断报道,但其具体的基因突变位点尚不清晰。现将本院2019年收治的1例GAMOS患儿的临床资料和基因测序结果报告如下,以提高对该疾病的认识。
患儿,女,1岁2个月,水肿、蛋白尿7 d。患儿出生后6个月会抬头,12个月会坐,但不能独坐,不会站立,语言发育迟缓,外院诊断为脑瘫。来院7 d前出现颜面部及双下肢水肿,当地医院查尿蛋白3+,血白蛋白18.1 g/L,肌酐17.7 μmol/L,尿素2.4 mmol/L,外院诊断为肾病综合征。
入院时查体发现血压76/45 mmHg,头围38 cm,胸围45 cm,体重8 kg,身高80 cm,特殊面容(鼻梁宽扁,眼间距过宽),语言发育迟缓,持续手足震颤,不能独坐。颜面部及双下肢水肿,腹部膨隆,移动性浊音呈阳性,双肺呼吸音粗,未闻及明显干、湿啰音,心脏查体未见异常。手指、脚趾细长,四肢肌张力减退。实验室检查存在以下特点:(1)低蛋白血症,高脂血症;(2)大量蛋白尿,肾小管损害;(3)免疫球蛋白水平低下;(4)内分泌检查未见异常;(5)肾脏超声未见异常。
外显子测序检查患儿和父母的基因,见表1。
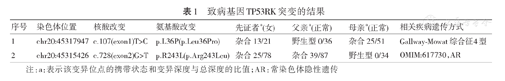
致病基因TP53RK突变的结果
致病基因TP53RK突变的结果
| 序号 | 染色体位置 | 核酸改变 | 氨基酸改变 | 先证者a(女) | 父亲a(正常) | 母亲a(正常) | 相关疾病遗传方式 |
|---|---|---|---|---|---|---|---|
| 1 | chr20:45317947 | c.107(exon1)T>C | p.L36P(p.Leu36Pro) | 杂合13/21 | 野生型0/36 | 杂合25/51 | Gallway-Mowat综合征4型 |
| 2 | chr20:45315426 | c.728(exon2)G>T | p.R243L(p.Arg243Leu) | 杂合25/78 | 杂合39/87 | 野生型0/34 | OMIM:617730,AR |
注:a:表示该变异位点的携带状态和变异深度与总深度的比值;AR:常染色体隐性遗传
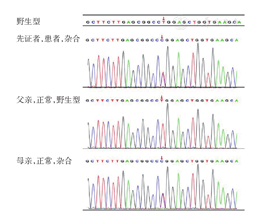
TP53RK基因突变1发生在第20号常染色体45317947位置的第107位碱基上。该处在正常情况下为胸腺嘧啶(T),先证者的父亲表型正常且基因型与野生型相同,而其母亲虽表型正常但基因发生改变,由T突变成胞嘧啶(C)并遗传给后代。先证者此处的碱基由T变异成C,使得其第36位氨基酸由亮氨酸(Leu)变为脯氨酸(Pro),为错义突变,该位点为T/C杂合,见图1。该突变在先证者上的变异深度与总深度的比值为13/21,其父亲为野生型,比值为0/36;而其母亲为杂合型,比值为25/51。
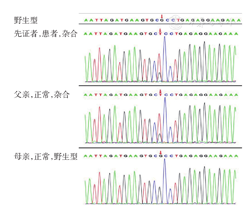
TP53RK基因突变2发生在第20号常染色体45315426位置2号外显子对应基因编码区的第728位碱基上。该处正常情况下为鸟嘌呤(G),先证者的母亲基因型和表现型均正常,而其父亲虽表型正常但基因发生变化,由G变异为T并遗传给先证者。先证者此处的碱基由G变异成T,使得其第243位氨基酸由精氨酸(Arg)变为亮氨酸(Leu),为错义突变,见图2,该位点为G/T杂合。在先证者上的变异深度与总深度的比值为25/78,其母亲为野生型,比值为0/34;其父亲为杂合型,比值为39/87。
根据美国遗传学及基因组学学会(ACMG)指南(2015年)[1],该研究中TP53RK基因的两个突变位点均位于深入研究的无良性变异外显子功能域,并且次要等位基因频率(minor allele frequency,MAF)小于0.005,这表明两个基因突变具有中等的致病概率。另外,通过保守性和蛋白结构预测软件预测出此变异对基因表达产物有影响,这提示两种基因突变可能致病。同时,该变异先证者为杂合子,符合AR病发病机制,且先证者及其家系成员表型及基因型符合分离定律。最重要的是,患者的临床表现符合TP53RK基因突变相关的GAMOS的部分临床特征,包括肾病综合征和全面性发育迟缓等,具有中等的临床特征匹配度。
综上所述,更正患儿诊断为:(1)GAMOS;(2)肾病综合征。
GAMOS是一种罕见的AR病,以肾病综合征合并小头畸形、智能和运动发育落后为主要临床特征[2]。自1968年Galloway和Mowat[3]首次报道两兄弟姐妹有早发肾病综合征、小头症和裂孔疝以来,目前已有超过60例GAMOS病例被报道。该病表型广泛,可伴视力和听力障碍、骨骼发育异常、手足畸形等;神经系统受损临床可表现为大脑、小脑及脑干发育不全,皮质萎缩,癫痫发作,精神活动性迟缓,智力发育迟缓等[4]。
此病常以肾病综合征起病,病理提示局灶节段硬化性肾小球肾炎居多,并迅速进展至终末期肾病,明确激素耐药,激素治疗只能短暂地延长寿命。如患儿发病较早,伴神经系统受累重者,无法进行肾移植,且常预后不佳,多数于6岁内死亡;如患儿肾病综合征出现较晚,其智能、运动发育落后相对较少,肾功能下降缓慢,激素或免疫抑制剂治疗有一定效果,该类患儿存活时间相对较长,甚至有15岁肾功能仍然正常的病例报道[5]。目前已开发了产前超声和围产期磁共振成像(MRI)应用于该病的临床诊断[6],但大多数报道的GAMOS病例仅能在出生后被诊断出来,产前检出的病例并不常见。
鉴于该疾病的临床异质性,越来越多的人考虑其发病原因可能具有遗传异质性。目前,由基因突变引起的GAMOS已在54个家庭中被报道,其中包括OSGEP突变26例(48.15%)、WDR73(Trp-Asp-repeat-containing protein 73)突变19例(35.18%)、TP53RK突变4例(7.40%)、LAGE3突变3例(5.55%)以及TPRKB突变2例(3.70%)[7]。
WDR73是一种WD重复蛋白,在维持细胞结构、存活和功能中扮演重要角色[8]。Colin等[9]最早报道了WDR73基因的缺失突变出现在2个不相关家族的GAMOS患者中,而且是第1个单基因突变引起的GAMOS。随后,Jinks等[8]在北美27例GAMOS患者中发现1个新的基因删除突变位点,佐证了Colin等[9]的研究结果。除此之外,由WDR73基因的纯合错义突变引起的GAMOS也被报道[10]。由WDR73基因突变引起的GAMOS患者常表现出严重的整体发育迟缓、迟发性肾病综合征、产后小头畸形、严重智力障碍及局灶阶段性肾小球硬化等肾脏病变[11]。
编码核孔蛋白107 KDa(NUP107)的基因变异也被发现与GAMOS的发生和进展相关。Miyake等[12]发现NUP107的双等位基因突变会导致早发性肾病综合征;而存在NUP107 p.Met101Ile的患者则显示局灶节段性肾小球硬化(FSGS)和整体发育迟缓[13]。Rosti等[14]在土耳其两个血亲家族的5例患者中发现NUP107 c.303G>A变异,临床表现包括小头畸形、肾病综合征和发育迟缓,与GAMOS的临床表现重合。与WDR73突变相比,NUP107突变引起的GAMOS患者一般没有脑结构缺损。由此推测NUP107在调节GAMOS患者的大脑生长方面扮演等位基因的角色。
最近1项大规模研究在患有GAMOS的37例患者(来自32个家族)中,发现了编码KEOPS亚基的OSGEP、TP53RK、TPRKB和LAGE3 4种基因中存在新的致病突变[15]。在该研究中,OSGEP突变最常见,在来自24个家族的28个个体中均被检测到。表型上,编码KEOPS亚基的4个基因任何一个发生突变都会引起患者出现原发性小头畸形、发育迟缓、癫痫发作倾向和早期发作的肾病综合征,大多数受影响的患者死于儿童早期[15]。
TP53RK的突变仅在来自3个家族的4个个体中检测到,各项病征的出现时间均较早。两例患者在2个月时出现早发性蛋白尿,在2.5月龄和11月龄时出现死亡和面部畸形;另外两例患者分别在1岁和10月龄时发病,后来分别在2.5岁和3岁时出现死亡[15]。另一韩国案例研究报道了与新型纯合TP53RK突变相关的家族性GAMOS病例(p.Lys65Met),突变类型为隐性突变,该家庭的4个孩子中有3个受到突变影响并表现出相似的表型,包括早发性肾病综合征、小头畸形、面部畸形和早期死亡[16]。
综上,本文报道了1例1岁2个月出现蛋白尿、语言发育迟缓、不能独坐以及特殊面容等症状的GAMOS患儿,对应肾病综合征、小脑发育不全和全面性发育迟缓、面部畸形等临床表型。与之前的报道不同,该患儿的TP53RK基因出现两处突变(c.107T>C和c.728G>T),且突变位点为首次发现。这将为该疾病在今后的临床诊断和鉴定中提供重要的指导意义。
所有作者均声明不存在利益冲突

























