
版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
患者 女,37岁,因"头晕、乏力2年余"入院。父母为非近亲结婚,无类似疾病家族史。查体:贫血貌,心肺无异常,肝肋下未及,脾脏肋下10 cm,质中等硬,边缘钝,无触痛。病理性神经反射未引出,智力正常。
实验室检查:血常规:白细胞1.64×109/L,血红蛋白85 g/L,血小板40×109/L。肝肾功正常。肝炎系列及自身抗体均阴性。胸部X光检查:心肺未见异常。B超:脾门处厚6.0 cm,脾长20 cm,实质回声均匀。骨髓细胞学形态:嗜碱染色细胞较易见,在涂片尾部可见尼曼-匹克细胞(图1A、图1B),细胞体积较大,不规则,直径20~80 μm,细胞质丰富,充满透明桑椹状脂肪滴,呈空泡状或泡沫状,细胞核较小,l~2个,常偏位,核染色质密集,这类细胞占全部有核细胞的1.6%。由于条件限制未能行酶学及基因学检测。
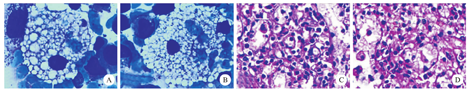
患者被诊断为尼曼-匹克病(Niemann-Pick disease,NPD)成人非神经型(E型)。入院后给予维生素C及E抗氧化治疗后效果不佳,查血常规:白细胞1.43×109/L,血红蛋白86 g/L,血小板43×109/L。于住院第15天在腹腔镜下脾脏切除术,手术顺利,术后无活动性出血,恢复良好。术后脾脏病理活检可见灶性的胞浆透亮泡沫样细胞(图1C、图1D),免疫组化:CD68(+),Ki67散在(+),S100(-),AACT(+),CD1a(-),CD31(+),符合NPD的诊断。脾脏病理活检确认患者为NPD。术后7天复查血常规:白细胞7.73 × 109/L,血红蛋白96 g/L,血小板534×109/L。术后1月复查血常规:白细胞4.64×109/L,血红蛋白115 g/L,血小板207×109/L。患者脾脏切除后血象逐渐恢复,治疗效果较好。
)
NPD又称鞘磷脂沉积病,是一种罕见的常染色体隐性遗传病,是由于神经鞘磷脂酶(acid sphingomyelinase,ASM)缺乏及活性降低所致,其发病率为(0.5~1.0)/10万,多见于中东、西欧、北美等地区。
NPD患者全身单核巨噬细胞系统被包含大量神经鞘磷脂的泡沫细胞浸润,全身各器官均有不同程度的神经鞘磷脂沉积,常伴有胆固醇增加,但血中神经鞘磷脂含量正常;肝、脾、淋巴结、骨髓、肺上皮细胞及神经节细胞呈空泡样。编码ASM蛋白的SMPD1基因突变是NPD主要的致病原因。骨髓或病理活检组织中见到尼曼-匹克细胞为诊断依据。测定神经鞘磷脂酶的活性、尿神经鞘磷脂酶排泄量及基因型分析可进一步确诊。Ries等[1]建议通过测定壳三糖苷酶的活性筛查早期诊断NPD,并发现该酶活性大于200 μmol/h·mL时诊断NPD敏感度为96%,特异度为100%。本例患者虽缺乏酶学及基因学检测,由于骨髓涂片及脾脏病理活检均可见尼曼-匹克细胞,故仍可确诊NPD。部分NPD患者可出现肾脏改变,但NPD病与肾脏改变并无直接的联系[2]。
据起病缓急和有无神经系统受累,可将本病分为A、B、C、D、E共5型,但也有介于A与B之间的类型的报道[3]。本例患者为成年女性,发病较晚,无神经系统症状,智力正常。结合年龄和临床资料分析,判断其属于成人非神经型(E型)。此型较为罕见,临床症状较轻,以脾肿大为最早的症状,不伴有神经系统异常,部分患者眼底黄斑部可见樱桃红斑点[4]。
NPD初诊时以全血细胞减少、脾大为主要表现,缺乏特异性,极易漏诊和误诊,应引起临床重视。近年来ASM活性和NPC-1基因检测为NPD的诊断提供了可靠的手段。不同类型的NPD神经鞘磷脂累积量及酶活性不同。A、B型患者ASM活性分别为正常值的5%~10%和5%~20%。C型患者该酶的活性为正常值的50%,而D、E型患者ASM活性正常。研究证实,NPD患者SMPD1基因第1外显子存在一个纯合突变T107C,使密码子由GTG变为GCG,导致36位缬氨酸被丙氨酸替代(p.V36A)。NPD患者多数较早死亡,死亡年龄为10~25岁。总体而言,神经系统症状出现越早,病情恶化就越快,越早死亡。
NPD无特效的治疗方法,临床上以对症治疗为主。可用维生素C、维生素E或丁羟基二苯乙烯阻止神经鞘磷脂所含不饱和脂肪酸的过氧化和聚合作用,减少脂褐素和自由基形成。动物实验显示,人绒毛膜上皮细胞移植和鞘磷脂酶靶向治疗等均有一定的疗效[5]。对于继发肝功能衰竭的B型NPD患者,肝移植可明显改善其症状和生活质量[6]。有报道用异基因造血干细胞移植成功治疗B型NPD并改善ASM水平[7]。成年人非神经型NPD合并脾功能亢进者可进行脾切除。本例患者入院后血常规三系明显降低,脾脏切除后血常规逐渐恢复正常,可为临床治疗该病提供新的探索。
无

























