
版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
例1 孕妇,24岁,G1P0,月经周期4/30天,自然怀孕。否认孕期放射线、毒物接触史,否认服用致畸药物。夫妻双方身体健康、无异常表型,非近亲婚配。孕24周行胎儿系统超声检查,发现胎儿左侧脑室后角增宽(10.1 mm),右侧脑室后角宽7 mm,第三脑室宽2.4 mm,第三脑室上方见10 mm×8.2 mm×6.6 mm液性暗区,无彩色血流信号,透明隔腔宽3.7 mm,提示胼胝体发育不良。胎儿头颅MRI显示两侧小脑半球尚清,后颅窝蛛网膜下腔宽径约0.57 cm,小脑蚓部存在,未见明确异常信号;胼胝体未见明确显示;双侧脑室分离,右侧脑室后角宽约1.0 cm,左侧脑室后角宽约1.1 cm,第三脑室宽约0.2 cm,上方可见一直径约8 mm液性间隙,第四脑室大小及形态未见异常;大脑各叶结构对称,未见明确异常信号影,中线结构无明显移位。诊断为胎儿胼胝体缺如。经遗传咨询及知情同意后,孕妇及家属选择终止妊娠。提取胎儿DNA进行染色体微阵列分析(chromosome microarray analysis,CMA),结果为arr 18q21.2q23(48 278 570-78 077 248)×1~2 (图1),提示染色体18q21.2-q23嵌合性缺失。缺失区域有171个基因,其中78个为OMIM收录基因,包括SMAD4、ATP8B1、CCBE1、CTDP1、CYB5A、DCC、FECH、MALT1、MC4R等。为获知该异常来源于父母遗传还是新发突变,对父母行外周血染色体检查,结果未见异常。
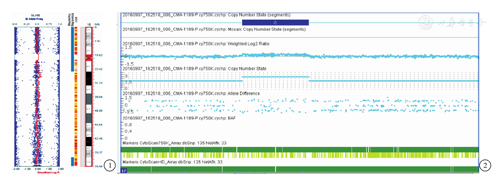
例2 患儿,男,4岁,剖宫产出生。出生体重3.2 kg,身长50 cm。2岁时行小儿智能发育量表(CDCC)测试,智能量表智力发展指数(Mental Developmental Index, MDI)<50(参考标准:MDI>80),相当于12.0月水平;动作量表精神运动发展指数(Psycho-motor Developmental Index, PDI)<50(参考标准:PDI>80),相当于9.7月水平。并且行0.35T MRI头颅平扫+弥散成像检查,结果显示两侧大脑半球对称,灰白质分界清晰,未见明显异常信号,枕大池大,胼胝体体部偏薄,两侧侧脑室体部平行,信号正常,第三、四脑室无扩张,中线结构居中无偏斜,脑干、小脑形态、信号未见明显异常,垂体信号可见,髓鞘化正常。弥散未见明显异常。局部筛窦以及左侧乳突区T2高信号。睡眠脑电图检查结果为相应年龄正常儿童脑电图。2岁半能独立行走,3岁多会喊爸爸妈妈。被诊断为脑瘫、智力低下。在我院就诊时,患儿母亲已妊娠19周,为避免再次生育类似患儿,要求进行遗传学检测,经知情同意后行CMA,结果为arr[hg19]17q11.2(29 061 631-30 395 625)×3(图2),涉及11个OMIM基因,包括NF1和RNF1。讨论 胼胝体是胎盘类哺乳动物脑内特有的神经纤维体,连接左右大脑半球,整合两半球之间的神经信息,是人类有效认知的功能基础,其发育情况可代表脑神经结构发育总体状况[1]。胼胝体发育不良(agenesis of corpus callosum,ACC)为先天性中枢神经系统畸形,1812年由Reil首次报道[2]。一般由影像学检查确诊,超声和MRI是直接可视的检查方式,但超声存在假阳性和假阴性。MRI是体内诊断ACC的金标准,并能区分部分性缺失和完全性缺失,同时可发现超声不能发现的其他颅脑结构异常[3,4,5]。根据是否伴发异常,分为单纯性(仅有胼胝体异常)和复合性(合并其他异常)ACC。目前病因仍未明确。我们通过CMA探索遗传因素在其发生过程中的作用,拓宽对此病的认识。
ACC的发生与环境、遗传相关。酒精综合征、代谢异常、胎儿宫内感染病毒都可导致其发生[3,6]。基因突变、缺失和重复均能影响胼胝体的形态、结构,从而产生一系列神经系统及神经系统外症状。在单纯性和复合性ACC患者中,文献统计染色体异常为18%。但最近研究显示,单纯性ACC患者罕有染色体异常,有学者认为染色体异常仅在复合性ACC中存在[7]。总体上,ACC的遗传异常主要体现在8、11、13-15、18号染色体上。基因缺失多表现在1、2、3、11、15、16、21、22和X染色体,缺失一个或多个基因。基因重复大多为4、5、6、8、11、12、14和19号染色体的部分三体;13、18、21、22号染色体的三体;以及三倍体等[8,9]。一些遗传综合征也与ACC的发生相关,常见的有X连锁显性遗传的Aicardi综合征、常染色体隐性遗传遗传的Andermann综合征。染色体的微缺失也与ACC相关,对此应做CMA[3]。
胼胝体发育涉及大量基因,基因异常可导致多种临床症状,目前很多基因和表型之间没有对应关系。本文例1胎儿缺失的DNA片段中包括多个致病基因,这些基因的异常可导致临床症状。其中RTTN突变导致胼胝体异常、智力缺陷、多小脑回畸形伴癫痫;CTDP1突变导致先天性白内障-面部畸形-神经病变综合征(congenital cataracts facial dysmorphism neuropathy syndrome,CCFDN);DCC突变导致Ⅰ型镜子动作;PIGN与Ⅰ型多发性先天异常-张力减退-癫痫综合征有关[10,11,12]。SMAD4、ATP8B1、CCBE1、CYB5A、FECH、MALT1、MC4R等基因的异常与其他一些疾病相关,疾病相关基因见表1。病例2患儿17q11.2微重复,有文献提示该片段重复可能与发育迟缓相关,但尚无确切的病因学依据[13]。该区域有致病基因NF1、RNF135。NF1突变与Ⅰ型神经纤维瘤、幼年型粒单核细胞白血病、沃森综合征、家族性脊髓神经纤维瘤、神经纤维瘤-努南综合征相关;而RNF135突变则与巨头-巨大儿-面部畸形综合征有关。
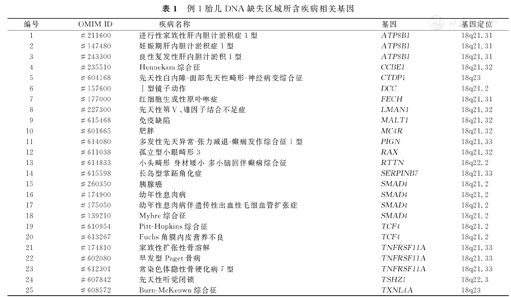
例1胎儿DNA缺失区域所含疾病相关基因
例1胎儿DNA缺失区域所含疾病相关基因
| 编号 | OMIM ID | 疾病名称 | 基因 | 基因定位 |
|---|---|---|---|---|
| 1 | #211600 | 进行性家族性肝内胆汁淤积症1型 | ATP8B1 | 18q21.31 |
| 2 | #147480 | 妊娠期肝内胆汁淤积症1型 | ATP8B1 | 18q21.31 |
| 3 | #243300 | 良性复发性肝内胆汁淤积1型 | ATP8B1 | 18q21.31 |
| 4 | #235510 | Hennekam综合征 | CCBE1 | 18q21.32 |
| 5 | #604168 | 先天性白内障-面部先天性畸形-神经病变综合征 | CTDP1 | 18q23 |
| 6 | #157600 | Ⅰ型镜子动作 | DCC | 18q21.2 |
| 7 | #177000 | 红细胞生成性原卟啉症 | FECH | 18q21.31 |
| 8 | #227300 | 先天性第Ⅴ、Ⅷ因子结合不足症 | LMAN1 | 18q21.32 |
| 9 | #615468 | 免疫缺陷 | MALT1 | 18q21.32 |
| 10 | #601665 | 肥胖 | MC4R | 18q21.32 |
| 11 | #614080 | 多发性先天异常-张力减退-癫痫发作综合征1型 | PIGN | 18q21.33 |
| 12 | #611038 | 孤立型小眼畸形3 | RAX | 18q21.32 |
| 13 | #614833 | 小头畸形身材矮小多小脑回伴癫痫综合征 | RTTN | 18q22.2 |
| 14 | #615598 | 长岛型掌跖角化症 | SERPINB7 | 18q21.33 |
| 15 | #260350 | 胰腺癌 | SMAD4 | 18q21.2 |
| 16 | #174900 | 幼年性息肉病 | SMAD4 | 18q21.2 |
| 17 | #175050 | 幼年性息肉病伴遗传性出血性毛细血管扩张症 | SMAD4 | 18q21.2 |
| 18 | #139210 | Myhre综合征 | SMAD4 | 18q21.2 |
| 19 | #610954 | Pitt-Hopkins综合征 | TCF4 | 18q21.2 |
| 20 | #613267 | Fuchs角膜内皮营养不良 | TCF4 | 18q21.2 |
| 21 | #174810 | 家族性扩张性骨溶解 | TNFRSF11A | 18q21.33 |
| 22 | #602080 | 早发型Paget骨病 | TNFRSF11A | 18q21.33 |
| 23 | #612301 | 常染色体隐性骨硬化病7型 | TNFRSF11A | 18q21.33 |
| 24 | #607842 | 先天性听觉闭锁 | TSHZ1 | 18q22.3 |
| 25 | #608572 | Burn-McKeown综合征 | TXNL4A | 18q23 |
ACC预后复杂、临床咨询棘手。患儿可伴有腭裂、头型异常、癫痫、发育迟缓、视听觉缺失等,新生儿期和童年期出现喂养困难的可能性高,并可存在如厕训练延迟、睡眠障碍、自伤行为、情绪失控、语言表达和接受障碍等问题,个体之间记忆力水平差异较大,智商水平随年龄增长有所改善。总的说来,大多研究认为ACC无特定预后,单纯性ACC大多预后良好,而复合性ACC通常预后不良。65%~75%单纯性ACC患者神经系统发育正常,但即便如此,单纯性ACC的预后也复杂多样;复合性ACC的预后取决于伴发的其他异常,如中枢神经系统异常、综合征及染色体异常等。在产前诊断过程中,有时很难确定ACC是否合并颅内、颅外异常、是否存在染色体异常,因此,建议对新诊断的ACC行CMA以获得较多的信息,便于咨询预后[1]。医生解释预后要严谨,因为目前大多关于ACC的研究、随访是短期的,对神经发育的观察局限在学前期,而大多患儿是在入学后表现出学习困难。远期表现如运动障碍、注意力缺陷多动障碍、自闭症等,需要到特定年龄才能观察到。另外,一些产前诊断为单纯性ACC的患儿在出生后发现其他异常。因此,产后及时的、长期的随访对最终诊断有重要的意义。
无

























