
随着基因检测技术的迅速发展,对于制定相应的标准和规范的需求也越来越迫切。临床遗传学检测报告是遗传检测过程中重要的一步,能够反映检测全过程中的质控问题。美国的政府机关、管理部门和行业协会等机构组织行业内专家制定了相应的标准指南,以规范或建议包括检测结果报告在内的整套临床遗传检测流程。美洲华人遗传学会组织特别工作组撰写本文,系统地介绍了美国临床遗传检测报告的要求和实施情况,并提供了范例与详尽的解释,希望有助于促进中国制定临床遗传检测报告的规范和标准。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
随着基因检测技术的快速发展,基因检测在临床上得到了广泛的应用。二代测序(next-generation sequencing,NGS)和微阵列芯片(chromosomal microarray,CMA)等高通量检测技术极大地提高了疾病的分子诊断水平,并为遗传性疾病和肿瘤的诊治及预防提供了可能。遗传检测报告是遗传学检测过程中的重要一步,不仅是临床医师进行遗传学诊断的依据,也是进一步进行临床干预以及指导下一代生育的依据。确保遗传检测的质量,不仅需要建立完善而规范的遗传检测认证和监督机构,更需要加强遗传检测过程的每个环节。临床遗传学检测报告能够反映检测过程各个环节的质量控制情况。因此,标准规范的检测报告可帮助临床遗传医师进行正确的诊断,并实施正确的治疗干预和产前诊断等,避免漏诊和误诊。
遗传检测报告的生成涉及到诸多环节和一系列专业规范化的过程,包括根据可靠的基因检测结果,结合患者的临床表型,依据相应的基因变异判定标准,对检测到的变异进行准确解释,并阐明变异的临床意义等。这一系列过程和环节均需按高度专业化的标准,并由有资质的人员来完成。美洲华人遗传学会组织专家工作组撰写本文,对美国的遗传检测报告规范标准和基本原则以及实施监督等方面进行系统的介绍,并附相应的检测报告模版供参考。
在美国,临床遗传检测分析技术和结果解读被界定为复杂且有高度技术要求的检测项目[1]。遗传检测标准规范涵盖了从检测分析的每一个步骤到检测报告发出的各个环节,包括临床遗传实验室的认证和监管、从业人员的认证和监管、以及实验检测分析技术的监管,具体参见《美国临床遗传检测实验室主任认证规范介绍》、《美国临床遗传实验员认证规范介绍》和《美国临床遗传检测实验室的标准及认证规范介绍》。针对分子检测,美国疾病控制中心提出实验室在实施过程中需有良好的行为习惯并严格执行[2]。临床遗传检测实验室须通过临床实验室改进修正案(Clinical Laboratory Improvement Amendments,CLIA)[3]以及所在州的认证机构的认可后,才有资格开展临床遗传检测项目。对实验室的认证许可将确保实验室能够严格遵守临床检测的标准和指导原则。这些标准规范,包括遗传检测报告的规范,通常由行业内的专家或组织机构制定。制定临床遗传检测标准规范的两个主要组织为美国医学遗传学和基因组学会(American Board of Medical Genetics and Genomics,ACMG)[4]和美国病理学家协会(College of American Pathologists,CAP)[5]。许多基因检测项目由实验室自行研发(Laboratory developed test,LDT)。这些检测尽管无需美国食品药品监督管理局(U.S. Food and Drug Administration,FDA)的批准,但任何实验室自行研发在被用于临床诊断之前,必须在实验室内进行反复的验证,以确保检测分析的特异性、灵敏度、准确性和精密度,使其具有可靠性和可重复性。因此,在检测报告中,必须注明实验室认证的具体情况以及检测技术和分析方法的局限性等。临床基因检测的每一个步骤都需要由具有相关知识技能和经验的专业人员来执行,检测结果也必须由经过认证的临床遗传学专家进行解读、签发,并在检测报告上签字。这些规范的执行情况则由国际医疗卫生机构认证联合委员会(The Joint Commission,TJC)[6]或CAP[5]等机构来监督。检测报告各部分的完成情况也包含在监督检查清单中。若违反规定,则可能导致某个检测项目被禁止,甚至实验室被关闭。因而,临床遗传检测报告的标准和规范是实验室总体标准和规范的一部分,与其他的标准规范互相补充,成为一整套的标准体系。
一份合格的临床遗传检测报告必须符合以下原则:
1.1 基因检测的结果必须准确无误。检测结果不仅对受检者本人,而且对其亲属的确诊、预后、治疗、干预、患病风险预测、预防、生育都有深远的指导意义。因此,在临床检测报告中所列出的每一项内容,从受检人的信息到检测结果,都必须准确无误。若发现错误,即使是错别字,也需要及时纠正并发出修正报告。
1.2 报告中所列的每项内容都必须定义明确且无歧义。遗传检测报告检出的变异,尤其胚系变异,对受检者而言是终生不变的。变异信息也是其亲属进行特定位点检测的依据。在报告发出后,许多相关的医师或部门均可能需要调看报告。因此,报告的内容必须清晰,避免模棱两可。例如,在美国,姓名顺序有各种写法。为使所列姓名无歧义,姓和名可以不同形式表示。可将姓为大写,名为小写,也可将姓列在前面,后面跟逗号,或在姓的下面划线,或将姓和名分别列出等,但实验室内部需采用统一的方法。为保证报告变异具有确定性,每个基因和变异均需采用国际公认的标准规则来命名。包括应注明参考基因组的版本、转录本的标号与版本等,以确保变异的命名具有唯一的可识别性。否则即使根据同一命名体系,也可能会导致引误解。
1.3 检测报告必须重点突出。应将最重要的结果和结论列在首页最醒目的位置,使阅读者能够一目了然。应清晰而有条理地标明报告中的每项内容和标题,方便临床医师根据需要对基本情况进行快速了解,并使他们容易找到更为详细的解释和其他相关信息。
1.4 检测结果的解读必须有充分的依据。在报告中,需详细解释基因与表型的关联性、变异的致病性、以及结果是否已能帮助达到送检的目的。需要列出判定变异结果的依据,包括依据何种指南、是否满足标准指南的条例、哪种证据和条件、对证据的解读分析等。若是依据文献,则需列出引用的文献出处。
1.5 检测报告须简明扼要。报告内容必须同检测的目的密切相关。对于无关内容的描述,不仅会淹没报告中的重要信息,还可能造成报告使用者或阅读者的误解和疑虑。因此,应避免无关的内容出现在报告中。所列的内容应针对受检者的实际情况,而不必面面俱到。对每位受检者可能都适用的有关背景和检测信息,可用网页链接的方式呈现,而不必在每份报告中详细列出。
1.6 适用范围和局限性必须标明。任何检测技术和方法都有其适用范围和局限性。检测报告需如实报道检测项目的目的、范围、方法、原理、质控参数、局限性等。列出这些指标,可帮助报告使用者根据报告来决定如何进行下一步的处理,包括采取诊断治疗或干预的措施、遗传咨询的方向和内容等。
临床遗传检测方法主要分为3种:临床细胞遗传学、临床分子遗传学和临床生化遗传学。这些方法都是临床遗传诊断的一部分,但又有各自独特的适用范围和侧重点。因此,检测报告的格式既有许多共性,又存在不少的差异(表1)。我们首先列出了遗传检测报告中的共同基本要素。各类报告的细节差异将在以后几节分别阐述。

临床遗传检测实验室的比较
临床遗传检测实验室的比较
| 实验室类型 | 临床生化遗传实验室 | 临床细胞遗传实验室 | 临床分子遗传实验室 |
|---|---|---|---|
| 主要方法 | 气/液相质谱 | 染色体核型、荧光原位杂交、芯片 | 测序、芯片、多重连接探针扩增、荧光原位杂交、定量PCR |
| 检测的异常类型 | 代谢物异常 | 染色体异常、基因位点异常、拷贝数变异 | 碱基序列变异、拷贝数变异 |
| 命名标准参考 | OMIM(www.omim.org) | 国际人类细胞遗传学命名委员会ISCN | 人类基因组变异命名委员会HGVS(http://varnomen.hgvs.org/) |
| 报告签署者认证资质 | ABMGG生化遗传学家 | ABMGG细胞遗传学家 | ABMGG分子遗传学家、分子病理医师 |
无论是哪种类型的临床遗传检测报告,都需要在实验室主任的监督下提供以下信息:
包括实验室的名称、地址、电话、传真号、网页地址、电子邮箱和实验室遵循的规范要求(如CLIA和CAP)、以及获得相应机构认证的编号和实验室负责人的姓名。通常在每份报告上都需要显示该实验室的独特徽标。
包括受检者的姓名、性别、出生日期或年龄、测试项目、恰当的临床信息、家族病史和(或)既往病史、受检者的唯一可识别编号和家系编号等。遗传测试的本质特点决定了家系信息至关重要。因此,必须在信息中包含家系编号。有些信息若受检者愿意提供,也可在报告中列出,如家庭地址、电话、传真号、电子邮箱等联系方式。
包括送检机构的名称、地址、科室等。在美国,患者在送检机构的医疗记录唯一编号(Medical Record Number,MRN)类似于中国的门诊号或住院号。若检测样本为送检机构内部实验室转送的样本,则还需有样本唯一编号,以便对标本进行唯一性识别。
包括开具检测医嘱且持有相应执照的临床医师(或其他合法授权人员)的姓名、联系地址、电话、传真号、电子邮箱等。实验室必须建立监管机制,以确保只有在授权人员的要求下才可对样本进行分析。在美国的一些州和其他国家,个人可在无医嘱的情况下选择进行一些实验室测试,但无论是哪种情况,报告中都要显示授权送检人的信息。
包括样本的唯一编号、样本来源、样本类型、样本采集时间、样本送达实验室的时间、样本处理的方法、样本质量评估等。
包括检测项目的范围、名称以及代号等。
包括结果摘要、结果的正确命名、综合分析结果和临床指征的相关性、所得结果是否能够满足临床需要依据该检测进行诊断的目的等。
包括对发现的基因变异或有临床意义的变异进行的判断和级别分类、对变异作出相应判断的依据、所遵循的标准与指南,或者对代谢物异常临床意义的解释。
包括在必要时可能需要提供遗传咨询的陈述,以及通过获得适当的临床数据或其他相关信息进一步明确遗传变异或代谢物异常的临床意义而提出的建议。
任何报告均应包括对检测技术方法的简要说明、检测平台的具体情况和分析所获得的数据和结果的方法的描述。
局限性说明包括由于方法和技术的限制可能造成的错检或漏检、以及无法检测到的异常情况等。若使用FDA批准的检测平台,则需要描述在该平台上进行测试验证的情况。若使用非FDA批准的检测平台,则需要描述在该平台上进行测试验证所使用的指标,并对测试的局限性进行声明。免责声明的内容包括可能导致的某些不明确的错误、偶然发现的异常、或使用非FDA批准的检测平台所造成的局限性等。
在对结果作出判定时,所用的证据若包括文献支持,则需列出所引用的文献。例如,在临床分子遗传检测中,应明确说明引用的文献中基因检测方法的有效性和实用性,详细说明科学证据基础。引用也包括针对所使用的检测,在解释结果时使用的数据库和文献。不列举报告中未引用的文献。
实验室主任或符合资格的指定人员须审查实验室报告的内容和格式,以确保能有效地传达患者的检测结果并满足临床的需求。经审核批准后,无论发出的检测报告为纸质版或电子版,均须由有认证资格的报告签发人员(表1)的签名和确切的签署时间。
生化遗传检测将根据特定的生化指标,判定结果是否在正常参考值范围内,用于指导遗传代谢病的诊治,是遗传检测的重要类别之一。因此,生化遗传检测报告除需要包含上述基本信息外,还需要根据专科的特点,提供生化指标的正常参考值范围,并判定结果是否为异常。对于异常的结果,在报告中要分析其临床意义与所提示的遗传病。若有可能,可推荐后续的其他检查,如酶活性测定、其他生化或分子检测等。有时检测指标的异常是某种遗传性疾病的诊断依据。然而,需要注意的是,在签发某种生化检测报告时,最好结合其他的检测结果和受检者的病史进行判断。如酰基肉碱检测结果报告可参考尿有机酸检测的结果。由于有些生化指标异常的原因并非遗传代谢病,而是样品污染、饮食、器官病变或抗生素使用,因此需要综合考虑,在排除假阳性的干扰后作出判断[7,8]。以下列举了3类特定的生化检测分析报告所应注意的事项:
酰基肉碱检测被广泛用于新生儿筛查和疑似患者的确诊。根据患者的年龄,有3个不同的酰基肉碱正常值范围。由于所使用的检测仪器和方法不同,加上人为误差,每个实验室应建立自己的正常值范围,而不能直接采用其他实验室的数值。另外,若在患者在未发病时采集样本,酰基肉碱的浓度可能为正常。因此,如果强烈怀疑某种疾病,可采用更为灵敏的办法,如尿酰基甘氨酸分析、基因测序等进行诊断。
新生儿肉碱的异常可能缘于母体。因此,在检测到新生儿血浆肉碱异常时,需检查母亲体内的肉碱含量和受检者尿液中的肉碱含量。此外,异常肉碱也可能是原发的或继发性的。需通过基因测序进一步确诊原发性的肉碱异常,或结合受检者病史判断其是否为继发性。
氨基酸检测样品可以来源于脑脊液、尿液或血浆,其中以血浆较为常用。不同的氨基酸异常组合可能与几种疾病相关,也可能仅提示唯一的疾病。检测报告需注明分析时所使用的检测仪器和方法(如氨基酸分析仪或液相色谱联用质谱仪)。由于在氨基酸分析中干扰因素较多,包括食物、抗生素、患者的身体状况等,有无禁食和采样时间都可能影响到氨基酸含量。在撰写报告时,需特别注意受检患者的具体情况。有时氨基酸检测需重复几次才能获得较为准确的结果。
尿有机酸检测能够非常有效地帮助临床确诊多种遗传代谢病。目前已有许多软件能够帮助确认检测到的化合物,但由于在气相色谱上不少化合物会迁移,或者软件在自动识别时会出错,因此每一份报告在经过软件辅助阅读后,需要人工进一步进行矫正。
染色体核型分析、荧光原位杂交(fluorescence in situ hybridization,FISH)和微阵列分析等细胞遗传学技术常被用于检测遗传性疾病和后天获得性肿瘤的染色体异常,且检测报告的内容和形式各有不同。我们在此将详细说明,除上述的共同基本要素外,在报告临床细胞遗传学检测的结果时的一些特点。美国没有全国统一的细胞遗传学临床实验室报告标准模板,我们在此附上若干不同类型的报告的例子(附件5~7)[9]。
目前细胞遗传学变异的命名与分类标准为国际人类细胞遗传学术语命名系统(Internation System of Chromosomal Nomenclature, ISCN)。为确保该命名系统能与技术的发展保持一致,国际细胞遗传学界提议由国际专家委员会对命名系统进行定期的审查和更新。该委员会由业界同行提名组成。目前已公布有1978、1981、1985、1991、1995、2005、2009、2013和2016共9个版本。
随着DNA测序技术在染色体异常检测中的应用日益广泛,国际人类细胞遗传学命名委员会和人类基因组变异协会(Human Genome Variation Society,HGVS)(表1)联合制定了ISCN2016最新版的命名标准,涵盖了通过染色体核型分析、FISH、微阵列芯片、特定染色体位点的分析(多重连接探针扩增技术、PCR等)以及DNA测序等多种技术能够检测到的细胞基因组变异的标准化描述[10]。因此,ISCN2016版被改称为国际人类细胞基因组遗传学术语命名系统,成为人类细胞遗传学家、分子遗传学家、实验室技术人员等不可或缺的参考书和在临床诊断中所使用的术语的通用标准。
对遗传性疾病的细胞遗传学检测报告需按ISCN2016制定的标准来命名,注明实验检测的方法,对阴性和阳性结果作出相关临床意义的解读,并说明该检测方法的局限性以及对患者和医师的进一步建议等。细胞遗传学的检测结论须放在报告最显著的位置,以便临床医师能够首先看到检测结果。可结合文献和参考书对结果进行判读和解读[11,12]。
在染色体核型分析中,对于检测方法的描述应包括细胞培养的数目、计数的细胞数目、进行染色体分析的细胞数目、用于制作核型的细胞数目以及染色体的分辨率等。染色体检测分析的分辨率为5~10 Mb。若染色体分析结果未见异常,则建议做染色体微阵列芯片或其他相关的遗传学检查,以排除更小范围的染色体变异。对于异常的染色体分析结果,需在报告中分析其临床意义。若能根据检测的结果对遗传病作出明确的诊断,则需对相关疾病的临床表现进行概述,并可考虑提出对相关家庭成员进行遗传检测和咨询的具体建议。
在FISH检测报告中,应同样按照ISCN的国际命名标准来描述检测的结果[10],注明检测方法,并说明FISH检测的局限性。例如,应注明该方法仅适用于检测相关基因位点,而无法检测到其他染色体的异常、嵌合体、小片段缺失、细微的染色体重排和相关基因的点突变等。若患者有明显的临床表现,则可在报告中建议使用其他方法进行进一步的检测。由于该方法属于对已知致病基因位点的检测,针对性较强。因此,若出现异常的检测结果,临床意义较为明确,则可结合医学文献,对遗传性疾病进行明确诊断。
在染色体微阵列芯片报告中,应对实验方法进行详细的描述,注明微阵列芯片的生产厂家、人类基因组的版本、对染色体片段缺失或重复的分辨率,并对其局限性进行特别说明,声明此方法可用于检测DNA拷贝数异常导致的缺失和重复,但不能检测拷贝数中性的染色体变化,如平衡易位、倒位或平衡插入等,无法可靠地检测< 10%的染色体嵌合,亦无法检测到未被标记物覆盖的染色体区域或DNA序列的变化,如点突变、小片段插入和缺失等。
对异常结果的解读和报告,应参见ACMG的标准和指南[13]。根据染色体缺失和重复的临床意义,将变异分为良性、疑似良性、意义未明、疑似致病和致病等5类。分类依据包括:(1)参考OMIM、GeneReviews、DECIPHER、HGMD等专业数据库和最新文献,判断缺失或重复是否会导致已知的遗传综合征或其他的特征性表型;(2)缺失或重复的大小;(3)是否为新发突变;(4)缺失或重复区域内所包含的基因及其致病机制;(5)参考内部和公开数据库,如基因组变异数据库(DGV)[14],比较变异在患者和普通人群的出现率。微阵列芯片目前被主要用于检测显性遗传病,需根据其分子机理判断变异是否可能致病。若某种显性遗传病是由于分子功能的增强而造成的,如检测到相关基因的缺失,则未必致病。某些基因缺失和重复可能导致不同的遗传病,在解读时须结合相关基因的分子机理进行判断。对于涉及隐性突变的常染色体微缺失,若与患者的症状不符,则属于意外发现,可按检测前知情同意的情况决定是否报告。对于X染色体连锁隐性遗传的致病性缺失通常需要报告。该染色体若被传递给男性后代,则可能导致症状。此外,部分女性携带者也可能出现不同程度的症状。染色体微缺失和重复区域中若未包含基因或无已知的致病基因且区域较小,可以不予报告。若缺失或重复在正常人群中的携带率高于疾病的发病率,且未见与疾病相关的报道,一般可判断为良性或疑似良性,不需要报告。若微阵列芯片检测发现疑似致病或致病变异,则需报告与此相关的遗传性疾病及其临床表现,并根据情况建议其亲属进行遗传检测和咨询。需要特别提醒的是,对染色体微缺失或重复的判断,由于认识的局限性和检测分析平台的不同,上述判定标准仅为一般规则。临床医师在诊断时,需根据检测报告,结合具体的临床表型作出判断。
在报告后天获得性肿瘤的体细胞变异时,对于结果和实验方法的描述应类似于遗传性疾病的报告格式。血液病和淋巴瘤体细胞变异检测的临床意义在于检测结果具有诊断和判断预后的价值。在这类报告中,通常不应对于如何治疗给予建议。在判断和解读结果时,可参考网络资料和有关书籍[15,16,17]。
临床细胞遗传学体细胞变异的检测样本大多采用肿瘤或疑似癌变的组织,在报告中应注明组织来源。若同一患者有多个样品,则报告中需说明所用样品的编号。体细胞变异检测样本常混有正常组织,且肿瘤具有异质性,通常仅在部分细胞中检测到变异,因此需要报告特定体细胞变异的比例。
对于染色体核型分析的实验方法的描述应包括细胞培养数目、计数的细胞数目、用于染色体核型分析的细胞数目、制作核型所用的细胞数目以及分辨率等。若异常染色体有临床意义,则应注明。例如,若染色体变异与急性早幼粒细胞白血病的诊断相符,检测结果对准确用药有指导意义,则提示预后较好。
体细胞变异的FISH检测在临床上的应用较为广泛,其优点是可对具有重要临床意义的变异进行快速检测。其检测报告与其他临床细胞遗传学报告的标准及内容类似,应依据ISCN国际标准报告所检测到的变异结果,并在报告中描述具体的实验检测方法和FISH检测的局限性等。对于异常的FISH检测结果,可结合病理报告和文献,对结果的临床意义进行分析和报告,并报告临床诊断或预后。
由于无需使用分裂细胞,染色体微阵列芯片技术能对全基因组进行高分辨率的检测,明确肿瘤组织内的染色体微缺失和微重复,因而具有重要的临床价值。在报告结果时,需依照ISCN国际标准,详细注明实验方法(包括微阵列芯片的生产厂家、人类基因组参考序列的版本,以及缺失或重复分辨率等)。在报告中,应注明检测方法的局限性。如附件所示,在B细胞急性淋巴细胞白血病的患者样本中,若检测到重要基因的缺失和融合基因,则提示预后较差,需要制定相应的治疗和随访方案。因此,对于具有临床意义的结果,需在报告中进行说明。
全基因组测序(Whole Genome Sequencing,WGS)是目前最全面和最复杂的分子遗传学检测方法。WGS不仅能够检测到小的序列变异,也能测到大片段的拷贝数变异和平衡的染色体结构变异,可以涵盖其他检测方法所检出的变异类型,其报告亦遵循相同的原则。此外,WGS不仅能够按医师的要求检测与受检者临床指征相关的基因,还可能检测出"次要发现"和"意外发现"。关于"次要发现"和"意外发现"的定义和区别,以及是否需要在报告中列出,请参见《美国临床基因检测前遗传咨询之要点》。我们以一份虚拟的WGS报告为例(附件8),介绍美国临床分子检测报告的标准规范。其中涉及的内容,包括患者信息、实验室信息、送检单位信息、各种编号、认证号、签名等均为虚构。描述的表型与变异数据则由作者根据真实的病例改编而成。
在临床分子遗传学检测报告中,碱基序列的参考标准为HGVS(表1)。碱基序列变异的分类标准参考ACMG/分子病理学协会(Association for Molecular Pathology,AMP)的指南[18]和中文翻译[19],此处不再赘述。
开展高通量测序的分子检测实验室必须符合相关的标准[20]。在检测报告中,应首先在显要位置醒目地给出总体结论。主要发现的结论可为"阳性"、"未决"或"阴性"。"阳性"是指检出的结果能够解释临床指征。例如,检出1个足以解释显性遗传病的致病/疑似致病变异,或足以解释常染色体隐性遗传病的双等位基因致病/疑似致病变异。"未决"是指检出的变异结果和临床指征相关,但仍无法完全解释临床指征。例如,仅在与表型相关的基因中检出了临床意义未明的变异,或仅检出1个与常染色体隐性遗传病相关的杂合致病/疑似致病变异。"阴性"是指未检出任何与临床指征相关的可能有临床意义的变异。若需报告次要和意外发现,结论可为"检出"或"未检出",而不建议写"阳性"或"阴性",并且只报告相关的致病/疑似致病变异。对经临床解读判断为良性和疑似良性的变异,不需要进行报告。对临床意义未明的变异,建议只在结论未决的情况下报告与受检者临床指征相关的变异。需要强调的是,对于报告的变异,均需要通过其他方法进行验证。例如,对于小的序列变异,可通过对PCR产物进行一代测序来验证。大片段拷贝数变异,可应用细胞遗传学芯片或FISH方法进行验证,也可应用分子遗传学多重连接探针扩增或定量PCR的方法验证。对染色体结构变异的断裂点,可通过对跨断点的PCR产物进行一代测序来验证。进行验证不只是为了排除假阳性,也是为了避免样品混淆及其他人为的错误。
在报告中,需对总体结论加以解释,包括基因与疾病的相关性、遗传模式、受检者的临床特征和家族史的符合度等,以说明检出的变异如何得出结论。并以列表的形式简明扼要地列出涉及的变异,其标准命名、参考序列、合子状态、相关疾病、遗传模式、致病性分类等。
之后,可根据情况给出建议,目的是为受检者建议后续的咨询服务,如何获得更多的信息,以阐明检测结果的意义。例如,可建议由临床遗传医师进行进一步的确诊并制定诊疗方案;建议受检者本人及家属寻求遗传咨询服务;建议患者进行其他生化、影像等临床检测,以及对其家庭成员进行遗传检测,以阐明临床意义未明的变异的影响等。但应避免直接提供临床诊断或干预方案,而是提醒医师结合临床作出判断。
在报告中,应该对每个变异作出详细的解读。用叙述性的语言给出变异分类评判的标准和依据,并用参考文献标明评判证据的来源。这些证据包括变异在患者和普通人群中的频率、变异相关的表型、遗传模式、是否为新发、生物信息工具对功能影响的预测、实验证实的功能影响等。此外,对基因变异相关的疾病应提供相关的背景知识,例如疾病的临床特征、遗传模式、分子机理、发病率、外显率等。其亲属的遗传风险也应在报告中说明。
若高通量测序的读长、深度和质量足够的话,在理论上将能够检出目标区域内的所有变异。但受技术设备和费用所限,目前实际获得的数据并不一定能够很好地覆盖所有的目标区域。尤其值得注意的是,"阴性结果"和"没有结果"两个概念并不等同。前者可排除病因,而后者若发生在与疾病高度相关的区域时,则提示需进行重复实验或用其他方法来弥补。在补充报告中,可列表说明与送检指征相关的特定基因/位点所达到的检测质控指标(如覆盖度)(附件9)。
基因检测已进入组学时代。生化遗传学检测中的代谢组学和蛋白组学、细胞遗传学检测中的全基因组染色体结构变异、分子遗传学检测中的全基因组测序等组学方法正逐渐被用于临床,并势必带来一些新的挑战和问题。
由于组学检测涉及的范围较广,而目前对于许多基因的认识仍十分有限,因此很多基因变异的临床意义仍无法确定。临床意义未明的变异有以下3种情况:(1)涉及的基因与受检者的临床指征相关,但由于当前信息不足,无法判断变异本身是否致病;(2)基因与表型的相关性尚不确定,但现有数据所提示的基因功能可能解释部分表型;(3)基因与表型的关系完全未知。对于这些临床意义未明的变异,是否需要报告应视具体情况而定。若检出的其他变异结论已达到检测的目的,则这类临床意义未明的变异无需报道。若未检出可解释临床指征的变异,则可报告第一类临床意义未明的变异,但必须注明为临床意义未明。报告这类变异的目的,是提示临床医师或其他相关人员进行进一步的临床表型相关性分析、家系的共分离分析等,以获得更多的证据,帮助判断基因和变异是否具有致病性。在其他情况下,临床意义未明的变异不必报告。
在受检者中检出的其他所有变异均可能对科研有用。因此,尽管在报告中不必列出这些变异,仍需提醒受检者若有需要,可向实验室索要报告中未列出的变异信息。
次要发现是指检测到与送检指征无关但对受检者有临床干预指导意义的变异。ACMG最新版的指南建议,报告与59个基因相关的次要发现[21]。意外发现则是指意外测到与受检者临床指征无关但有遗传意义的基因变异,比如受检者携带一些常见的严重遗传病的基因变异。对于次要发现和意外发现,是否需要报告应取决于受检者的意愿。由于基因组学检测的复杂性和检测的广泛性,必须经过检测前的遗传咨询,让受检者详细了解组学检测的利弊,进行知情同意选择,具体参见《美国临床基因检测前遗传咨询之要点》。若受检者要求得知次要发现或意外发现,临床实验室则需出具所涉及的致病或疑似致病变异的报告(附件10)。
美国联邦法律规定,受检者有权获得自身临床检测的原始数据[22]。一般情况下,实验室仅在受检者提出返还要求后才返还其数据。受检者可索取的数据包括,在受检者提出要求时,临床实验室所保存的与受检者隐私相关的所有数据。受检者的健康信息,包括原始基因检测数据,属于受检者的隐私,其所有权应归受检者。临床实验室有返还数据的义务。美国各州均制定了法律,规定临床实验室必须在一定时期内保留受检者重要的遗传检测数据。一般来说,保留期为一代人的时间(20年)。
随着基因检测技术的不断发展,人们对基因的认识日新月异,对于原始基因数据的解读能力也在迅速增强。然而,对数据进行重新分析和解读需要一定的人力和物力。实验室可根据自身的资源和可行性决定是否进行重新解读或重新解读的间隔时间。目前美国对此尚无明文规定。各实验室可根据自身的情况,制定相应的标准操作流程,之后严格执行。例如,有些实验室规定,当遇到意义未明的变异时,需进行重新解读和分类。若解读的结果与之前发出的报告有所不同,则需对既往涉及相同变异的所有案例发布修正报告。也有实验室规定,应每隔一段时间(如半年或1年)对未决和阴性的案例进行重新分析。
遗传检测报告的质量是遗传检测过程所有环节的规范标准的集中体现。本文详细介绍了美国各种遗传检测报告的标准。根据其侧重的领域,现有的临床遗传学检测又分为生化遗传检测、细胞遗传学检测和分子遗传学检测。其中,受检者和实验室的基本信息在不同亚专业的报告中并无太大的差别。总体来说,涵盖的基本信息量越多,就越便于对检测样本进行回溯和唯一性确认。在生化遗传检测报告中,应强调需要根据检测到的特定生化指标,结合患者的临床表现作出相应的诊断。实验室需建立相应人群的正常值范围并在报告中列出。生化指标的检测结果与患者的饮食、取样时间、疾病状态、用药情况等密切相关。因此,提供和说明这些信息将有助于准确解读结果。在细胞遗传学检测报告中,应根据国际命名术语对染色体异常进行描述,并附染色体图谱供临床医师参考。染色体微阵列芯片检测报告可根据现有的指南进行解读和分析[13]。但需要注意的是,由于检测平台与方法、以及检测过程与数据分析的不同,不同的实验室对于结果的解读可能存在很大的差异。因此,各实验室需详细列出检测平台和方法、分析工具和质控参数。检测到的结果变异需标明参考标注的版本信息、变异的范围以及涵盖的基因等。在分子检测报告中,尽管ACMG/AMP发布了DNA序列变异的分类指南[18]且已被译成中文[19],但指南所列仅为普遍原则,而具体条款的细节仍需按疾病与基因的特点进行细化,并随着对于基因型和表型认知的提升不断修正。高通量的基因检测将可能产生次要和意外发现,如何进行报告应根据受检者知情同意的选择。对于原始数据的返还,美国强调保护患者对自身基因数据的拥有权。我们详尽介绍了美国临床遗传学检测报告的要点和标准,期望国内同行能结合实际情况制定相应的规范和标准,以进一步提高临床遗传学检测的质量,促进临床遗传学的规范化发展。
所有作者均声明不存在利益及版权冲突

酰肉碱检测报告
检测项目:酰肉碱检测
受检者临床指征:生长发育迟缓
检测结论:正常
检测结果在正常参考范围内
检测结果
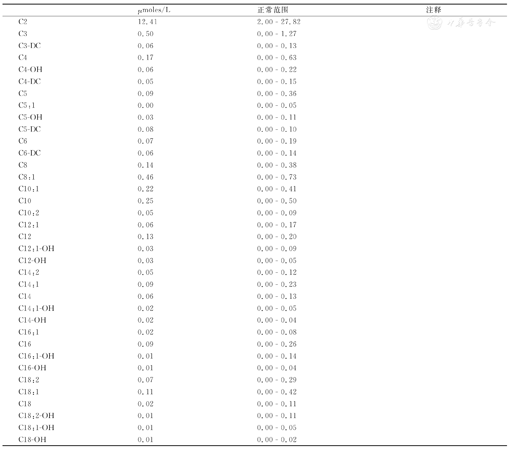
| μmoles/L | 正常范围 | 注释 | |
|---|---|---|---|
| C2 | 12.41 | 2.00 - 27.82 | |
| C3 | 0.50 | 0.00 - 1.27 | |
| C3-DC | 0.06 | 0.00 - 0.13 | |
| C4 | 0.17 | 0.00 - 0.63 | |
| C4-OH | 0.06 | 0.00 - 0.22 | |
| C4-DC | 0.05 | 0.00 - 0.15 | |
| C5 | 0.09 | 0.00 - 0.36 | |
| C5:1 | 0.00 | 0.00 - 0.05 | |
| C5-OH | 0.03 | 0.00 - 0.11 | |
| C5-DC | 0.08 | 0.00 - 0.10 | |
| C6 | 0.07 | 0.00 - 0.19 | |
| C6-DC | 0.06 | 0.00 - 0.14 | |
| C8 | 0.14 | 0.00 - 0.38 | |
| C8:1 | 0.46 | 0.00 - 0.73 | |
| C10:1 | 0.22 | 0.00 - 0.41 | |
| C10 | 0.25 | 0.00 - 0.50 | |
| C10:2 | 0.05 | 0.00 - 0.09 | |
| C12:1 | 0.06 | 0.00 - 0.17 | |
| C12 | 0.13 | 0.00 - 0.20 | |
| C12:1-OH | 0.03 | 0.00 - 0.09 | |
| C12-OH | 0.03 | 0.00 - 0.05 | |
| C14:2 | 0.05 | 0.00 - 0.12 | |
| C14:1 | 0.09 | 0.00 - 0.23 | |
| C14 | 0.06 | 0.00 - 0.13 | |
| C14:1-OH | 0.02 | 0.00 - 0.05 | |
| C14-OH | 0.02 | 0.00 - 0.04 | |
| C16:1 | 0.02 | 0.00 - 0.08 | |
| C16 | 0.09 | 0.00 - 0.26 | |
| C16:1-OH | 0.01 | 0.00 - 0.14 | |
| C16-OH | 0.01 | 0.00 - 0.04 | |
| C18:2 | 0.07 | 0.00 - 0.29 | |
| C18:1 | 0.11 | 0.00 - 0.42 | |
| C18 | 0.02 | 0.00 - 0.11 | |
| C18:2-OH | 0.01 | 0.00 - 0.11 | |
| C18:1-OH | 0.01 | 0.00 - 0.05 | |
| C18-OH | 0.01 | 0.00 - 0.02 |
检测结果解释
检测结果在正常参考范围内
建议
该检测结果需结合受检者的临床表现等进行综合分析,必要时复查。
关于本报告的其他问题或索取当地健康咨询服务,请联系DNA实验室,电邮:info@dnalaboratory.com或致电:(123) 456-7890。
检测方法
应用液相色谱质谱联用仪检测酰肉碱
局限性
某些有机酸病用该检测方法不易检测到,如维生素B12代谢病。某些酰肉碱异常有时只在受检者发病时才能检测到。
声明
本报告结果不代表最终诊断结果,仅供临床医生参考。实验结果和解释受局限于收集的样品、实验方法和现有知识,临床医生需结合受检者的具体情况,利用报告的结果和注释,作出临床判断。
报告:XX,医学博士,2018年3月20日上午10:41
审核:XX,医学博士,2018年3月20日下午12:30
电子签名:XX,医学博士,2018年3月20日下午12:30

肉碱检测报告
检测项目:血清肉碱检测
受检者临床指征:生长发育迟缓
结论:异常
检测结果与患者临床表现相符合
检测结果

| 肉碱 | 代号 | 结果 | 注释 | 参考值 | 单位 |
|---|---|---|---|---|---|
| 总肉碱 | TC | 10 | 低 | 17 - 41 | nmol/mL |
| 游离肉碱 | FC | 2 | 低 | 10 - 21 | nmol/mL |
| 酰肉碱(TC-FC) | AC | 8 | 3 - 24 | nmol/mL | |
| 酰肉碱/游离肉碱(AC/FC)比例 | AC/FC | 4.0 | 0.2 - 1.4 | - |
检测结果解释
游离肉碱低于正常参考值,可能是由于原发性肉碱缺乏症或者是次发性的结果。
建议
该结果需要结合受检者的临床表现等进行综合分析。建议对该受检者进一步进行尿肉碱检测和CTP1基因分子基因检测,检测其母亲血清肉碱等。建议该受检者到临床医师处进行定期随访及复查。关于本报告的其他问题或索取当地健康咨询服务,请联系DNA实验室,电邮:info@dnalaboratory.com或致电:(123) 456-7890。
检测方法
应用液相色谱质谱联用仪检测肉碱含量。
局限性
实验结果和解释受局限于收集的样品、实验方法和现有知识。
声明
本报告结果不代表最终诊断结果,仅供临床医生参考。临床医生需结合受检者的具体情况,利用报告的结果和注释,作出临床判断。
报告:XX,医学博士,2018年3月20日上午10:41
审核:XX,医学博士,2018年3月20日下午12:30
电子签名:XX,医学博士,2018年3月20日下午12:30

氨基酸检测报告
检测项目:氨基酸检测
受检者临床指征:枫糖尿病患者随访
结论:异常
检测结果与患者临床表现相符合
检测结果
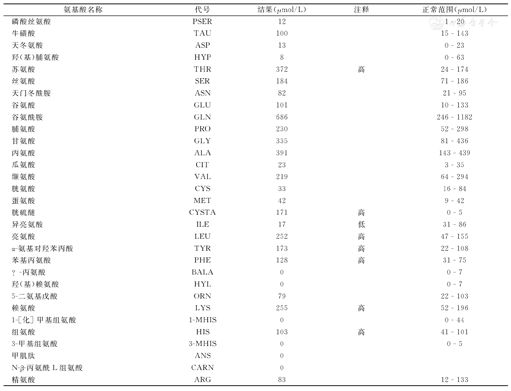
| 氨基酸名称 | 代号 | 结果(μmol/L) | 注释 | 正常范围(μmol/L) |
|---|---|---|---|---|
| 磷酸丝氨酸 | PSER | 12 | 1 - 20 | |
| 牛磺酸 | TAU | 100 | 15 - 143 | |
| 天冬氨酸 | ASP | 13 | 0 - 23 | |
| 羟(基)脯氨酸 | HYP | 8 | 0 - 63 | |
| 苏氨酸 | THR | 372 | 高 | 24 - 174 |
| 丝氨酸 | SER | 184 | 71 - 186 | |
| 天门冬酰胺 | ASN | 82 | 21 - 95 | |
| 谷氨酸 | GLU | 101 | 10 - 133 | |
| 谷氨酰胺 | GLN | 686 | 246 - 1182 | |
| 脯氨酸 | PRO | 230 | 52 - 298 | |
| 甘氨酸 | GLY | 335 | 81 - 436 | |
| 丙氨酸 | ALA | 391 | 143 - 439 | |
| 瓜氨酸 | CIT | 23 | 3 - 35 | |
| 缬氨酸 | VAL | 219 | 64 - 294 | |
| 胱氨酸 | CYS | 33 | 16 - 84 | |
| 蛋氨酸 | MET | 42 | 9 - 42 | |
| 胱硫醚 | CYSTA | 171 | 高 | 0 - 5 |
| 异亮氨酸 | ILE | 17 | 低 | 31 - 86 |
| 亮氨酸 | LEU | 252 | 高 | 47 - 155 |
| α-氨基对羟苯丙酸 | TYR | 173 | 高 | 22 - 108 |
| 苯基丙氨酸 | PHE | 128 | 高 | 31 - 75 |
| ?-丙氨酸 | BALA | 0 | 0 - 7 | |
| 羟(基)赖氨酸 | HYL | 0 | 0 - 7 | |
| 5-二氨基戊酸 | ORN | 79 | 22 - 103 | |
| 赖氨酸 | LYS | 255 | 高 | 52 - 196 |
| 1-[化]甲基组氨酸 | 1-MHIS | 0 | 0 - 44 | |
| 组氨酸 | HIS | 103 | 高 | 41 - 101 |
| 3-甲基组氨酸 | 3-MHIS | 0 | 0 - 5 | |
| 甲肌肽 | ANS | 0 | ||
| N-β-丙氨酰L组氨酸 | CARN | 0 | ||
| 精氨酸 | ARG | 83 | 12 - 133 |
检测结果解释
受检者为已确诊的枫糖尿病患者,目前正在接受临床治疗。亮氨酸252μmol/L落在目标治疗区间(155至300),结果显示患者的饮食治疗效果良好。其他氨基酸的异常可能为未进行禁食造成的结果。需注意,胱硫醚和异亮氨酸在此结果图谱中有重合现象。
建议
该结果需结合患者的临床表现等进行综合分析。建议该患者到临床医师处接受定期随访及复查。关于本报告的其他问题或索取当地健康咨询服务,请联系DNA实验室,电邮:info@dnalaboratory.com或致电:(123) 456-7890。
检测方法
应用Biochrom氨基酸分析仪检测血液中氨基酸含量。
局限性
因为在氨基酸分析中干扰因素较多,包括食物、抗生素、患者的身体状况等,所以有无禁食和采样时间都可能影响到氨基酸含量。实验结果和解释受局限于收集的样品、实验方法和现有知识。
声明
本报告结果不代表最终诊断结果,仅供临床医生参考。临床医生需结合受检者的具体情况,利用报告的结果和注释,作出临床判断。
报告:XX,医学博士,2018年3月20日上午10:41
审核:XX,医学博士,2018年3月20日下午12:30
电子签名:XX,医学博士,2018年3月20日下午12:30

有机酸检测报告
检测项目:有机酸检测
受检者临床指征:生长发育迟缓
检测结论:异常
检测结果与戊二酸血症I型相符合
检测结果解释
患者尿液戊二酸和3-羟基戊二酸显著升高,结合血液内C5-dc酰肉碱升高,该患者实验室检测符合戊二酸血症I型的特征,但仍需临床医师结合患者的临床表现进行明确诊断。
建议
该结果需结合患者的临床表现等进行综合分析。建议该患者到临床医师处进行定期随访及复查。关于本报告的其他问题或索取当地健康咨询服务,请联系DNA实验室,电邮:info@dnalaboratory.com或致电:(123) 456-7890。
检测方法
应用气相色谱仪检测尿液中有机酸含量。
局限性
部分遗传代谢病,如尿素循环、脂肪酸氧化缺陷、生物素代谢缺陷等无法通过检测尿液中有机酸加以排除。若收集样品时受检者处于未发病状态,也可能无法检测到。实验结果和解释受局限于收集的样品、实验方法和现有知识。
声明
本报告结果不代表最终诊断结果,仅供临床医生参考。临床医生需结合受检者的具体情况,利用报告的结果和注释,作出临床判断。
报告:XX,医学博士,2018年3月20日上午10:41
审核:XX,医学博士,2018年3月20日下午12:30
电子签名:XX,医学博士,2018年3月20日下午12:30

遗传性疾病染色体检测报告
结果:阴性
ISCN核型:46,XY
结果解释:
正常男性,在本检测中应用的染色体条带分辨率下未观察到临床上有意义的染色体数目或结构异常。
局限性:
正常的染色体结果不排除可能引起该受检者临床表现的其他细胞遗传学改变,包括微小的染色体变异或极低水平的嵌合体。
建议:
必要时可应用微阵列芯片检测,以排除G带染色体分析无法检测到的缺失或重复。该检测的相关信息可在我们的实验室网站cytolaboratory.com找到。此外,应考虑进行完整的遗传评估,以排除与该患者临床表现相关的其他遗传病因。
检测方法:染色体核型分析
细胞培养数目:2
计数的细胞数目:20
染色体分析的细胞数目:5
制作核型的细胞数目:2
染色体条带分辨率:550
声明:
1.报告结果只对本次送检样本负责。
2.关于本报告的其他问题或需要进行相关健康咨询服务,请联系检测实验室。
电子邮箱:info@dnalaboratory.com 联系电话:(123) 456-7890
报告:XX,医学博士,2018年3月20日上午10:41
审核:XX,医学博士,2018年3月20日下午12:30
电子签名:XX,医学博士,2018年3月20日下午12:30

遗传性疾病染色体检测报告
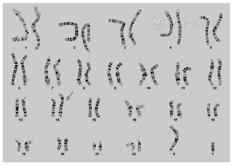
结果:阳性
ISCN核型:46,XY,der(14;21)(q10;q10),+21
结果解释:
异常染色体核型,符合唐氏综合征,为14号染色体和21号染色体间的不平衡罗伯逊易位所致。
分析的处于分裂中期的所有细胞均含有46条染色体,包含14号染色体长臂和21号染色体长臂的易位染色体。因这些细胞均含有一个正常14号染色体,两个正常21号染色体,以及易位或衍生的染色体(以"der"表示)。该核型为21号染色体三体。
该核型结果与唐氏综合征的临床诊断一致[MIM#190685]。唐氏综合征是新生儿最常见的染色体异常之一,临床表现明确,包括特殊面部特征、轻到中度智力障碍,可有先天性心脏病、胃肠道畸形和传导性听力障碍等。
建议:
因存在易位染色体,建议进一步检测父母的染色体核型,以确定他们是否为易位平衡携带者。具体产前遗传咨询或处理,请到临床医师处就诊或咨询。
检测方法:
细胞培养数目:2
计数的细胞数目:20
染色体分析的细胞数目:5
制作核型的细胞数目:2
染色体条带分辨率:550
局限性:该结果不排除可能引起该受检者临床表现的其他细胞遗传改变,包括微小的染色体变异或极低水平的嵌合体。
声明:
1.报告结果只对本次送检样本负责。
2.关于本报告的其他问题或需要进行相关健康咨询服务,请联系检测实验室。
电子邮箱:info@dnalaboratory.com 联系电话:(123) 456-7890
报告:XX,医学博士,2018年3月20日上午10:41
审核:XX,医学博士,2018年3月20日下午12:30
电子签名:XX,医学博士,2018年3月20日下午12:30

肿瘤细胞体细胞变异的染色体核型分析报告
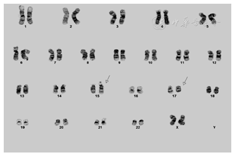
结果:阳性
ISCN核型:46,XX,t(15;17)(q24;q21)[20]
结果解释:异常染色体核型,t(15; 17)(q24; q21)/ PML-RARA平衡易位。结合已提供的临床信息和荧光原位杂交检测(FISH)结果,与急性早幼粒细胞白血病的诊断相符合。
在分析的20个GTW染色的处于分裂中期的细胞,均观察到异常核型。急性早幼粒细胞白血病(APL)常常与播散性血管内凝血有关。应用最佳治疗方案,t(15; 17)阳性急性早幼粒细胞白血病预后好,治愈率高。
建议:至临床医师进行就诊,结合其他临床数据和FISH结果进行明确诊断,并进行定期实验随访。
检测方法:
细胞培养:24小时无刺激培养,检测观察快速分裂细胞的异常。并进行48小时无刺激培养,检测观察较慢分裂细胞中的异常。
计数的细胞数目:20
分析的细胞数目:20
制作核型的细胞数目:2
染色体条带分辨率:350-400
局限性:该结果不排除微小的或极低水平的染色体变异。
声明:
1.报告结果只对本次送检样本负责。
2.关于本报告的其他问题或需要进行相关健康咨询服务,请联系检测实验室。
电子邮箱:info@dnalaboratory.com 联系电话:(123) 456-7890
报告:XX,医学博士,2018年3月20日上午10:41
审核:XX,医学博士,2018年3月20日下午12:30
电子签名:XX,医学博士,2018年3月20日下午12:30

遗传性疾病荧光原位杂交检测报告
结果:阳性
检测结论:
RAI1基因缺失。此缺失导致史密斯-马吉利综合征(Smith-Magenis syndrome,OMIM:# 182290)。
检测结果:
ISCN: ish del(17)(p11.2p11.2)(RAI1-)
结果解释:
中期分裂相细胞内的一条17号染色体无绿色信号(RAI1),表明RAI1基因缺失。正常17号染色体显示绿色和红色信号(对照位点)。
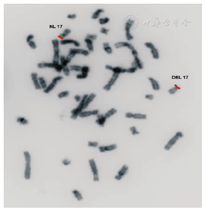
检测方法:
此检测应用RAI1/XXXX双色探针(探针来源:XXXX公司),分析10个以上中期细胞。
局限性:
此分析仅用于对上述基因位点进行检测,并不能检测到其他染色体异常。亦无法用于检测嵌合体,染色体小片段缺失,细微的染色体重排和RAI1基因的点突变。
声明:
1.报告结果只对本次送检样本负责。
2.关于本报告的其他问题或需要进行相关健康咨询服务,请联系检测实验室。
电子邮箱:info@dnalaboratory.com 联系电话:(123) 456-7890
报告:XX,医学博士,2018年3月20日上午10:41
审核:XX,医学博士,2018年3月20日下午12:30
电子签名:XX,医学博士,2018年3月20日下午12:30

遗传性疾病荧光原位杂交检测报告
结果:阴性
检测结论:
未见RAI1基因缺失。
检验结果:
ISCN:ish 17p11.2(RAI1×2)
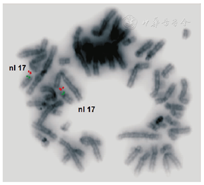
结果解释:
中期分裂相细胞内的两条17号染色体均出现绿色信号(RAI1)和红色信号(对照位点),未见RAI1基因缺失。
检测方法:
应用RAI1/XXXX双色探针(探针来源:XXXX公司),分析10个以上的中期细胞。
局限性:
此分析仅用于检测上述基因位点,并不检测其他染色体异常。亦无法用于检测嵌合体,小片段染色体缺失,细微的染色体重排和RAI1基因的点突变。
声明:
1.报告结果只对本次送检样本负责。
2.关于本报告的其他问题或需要进行相关健康咨询服务,请联系检测实验室。
电子邮箱:info@dnalaboratory.com 联系电话:(123) 456-7890
报告:XX,医学博士,2018年3月20日上午10:41
审核:XX,医学博士,2018年3月20日下午12:30
电子签名:XX,医学博士,2018年3月20日下午12:30

肿瘤体细胞变异的荧光原位杂交检测报告
结果:阳性
检测结论:
PML/RARA融合基因阳性。符合急性早幼粒细胞白血病(APL)的诊断。
检验结果:
ISCN: nuc ish(PML,RARA)×3(PML con RARA×2)[152/200]
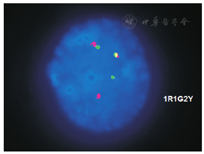
检测方法:
此检测应用PML/RARA双色探针(探针来源:XXXX公司),检测PML/RARA融合基因的特异性信号。
结果解释:
76% (152/200)的间期分裂相细胞显示一个红色信号(PML),一个绿色信号(RARA)和两个黄色融合信号(PML/RARA),表明PML/RARA融合基因为阳性。该融合基因通常由15号和17号染色体相互易位导致。关于染色体核型检验结果见另外单独报告。
局限性:
此检测方法仅用于检测上述基因位点,并不能检测到其他染色体异常。且可检测的阀值为阳性间期分裂相细胞不低于xx%。
声明:
1.报告结果只对本次送检样本负责。
2.关于本报告的其他问题或需要进行相关健康咨询服务,请联系检测实验室。
电子邮箱:info@dnalaboratory.com 联系电话:(123) 456-7890
报告:XX,医学博士,2018年3月20日上午10:41
审核:XX,医学博士,2018年3月20日下午12:30
电子签名:XX,医学博士,2018年3月20日下午12:30

肿瘤组织体细胞变异的荧光原位杂交检测报告
结果:阴性
检测结论:
未检测到PML/RARA融合基因。
检验结果:
ISCN: nuc ish(PML,RARA)×2[200]
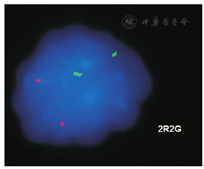
检测方法:
此检测应用PML/RARA双色探针(探针来源:XXXX公司),检测PML/RARA融合基因的特异性信号。
结果解释:
间期分裂相细胞显示两个红色信号(PML)和两个绿色信号(RARA),未显示融合基因(PML/RARA)的异常信号。
建议:
如患者有明显的APL临床表现,请至临床医师处就诊。建议应用PCR方法检测PML/RARA融合基因。染色体核型分析结果另见单独报告。
局限性:
此分析仅用于检测上述基因位点,并不检测其他染色体异常。检测的阀值为阳性间期分裂相细胞不低于xx%。大约2-5%的急性早幼粒细胞白血病(APL)患者有RARA插入PML导致的罕见融合基因,应用该方法也不能检测到。
声明:
1.报告结果只对本次送检样本负责。
2.关于本报告的其他问题或需要进行相关健康咨询服务,请联系检测实验室。
电子邮箱:info@dnalaboratory.com 联系电话:(123) 456-7890
报告:XX,医学博士,2018年3月20日上午10:41
审核:XX,医学博士,2018年3月20日下午12:30
电子签名:XX,医学博士,2018年3月20日下午12:30

遗传性疾病染色体微阵列芯片检测报告
结果:阴性
检测结论:无异常
检测结果:
ISCN:arr(1-22)×2,(X,Y)×1
检测结果解释:
未检测目前已知的染色体微缺失或重复疾病相关的拷贝数变化
检验方法:
将患者的DNA和同性别的正常对照DNA杂交到XXXXXXXX芯片(芯片版本CMAXXXX,芯片编号XXXXXXXXXXXX-X),分析杂交信号,检测受检样本的染色体拷贝数异常情况。此芯片在临床疾病相关的染色体区域的平均分辨率为xx kb,可检测到的最小缺失和扩增为xx kb。数据分析和解析依据本实验室的内部数据库和公众数据库,包括UCSC Genome Browser,Database of Genomic Variants (DGV),Mutation Database (HGMD),DECIPHER以及科学文献。我们对于小于300 kb的无已知表型的拷贝数变化将酌情根据具体情况决定是否给予报告。
局限性:
此方法仅用于检测拷贝数异常导致的缺失和重复,无法检测到拷贝数正常的染色体变化,如平衡易位,倒位或平衡插入。此方法亦无法可靠地检测低于10%的染色体嵌合体,亦无法检测探针未覆盖的染色体区域。也不能用于DNA序列的变化,如点突变,小片段插入和缺失等的检测。
声明:
1.报告结果只对本次送检样本负责。
2.关于本报告的其他问题或需要进行相关健康咨询服务,请联系检测实验室。
电子邮箱:info@dnalaboratory.com 联系电话:(123) 456-7890
报告:XX,医学博士,2018年3月20日上午10:41
审核:XX,医学博士,2018年3月20日下午12:30
电子签名:XX,医学博士,2018年3月20日下午12:30

遗传性疾病染色体微阵列芯片检测报告
结果:阳性
检测结论:异常-染色体片段缺失,22q11.2缺失综合征
检验结果:arr[GRCh37] 22q11.21(18918741_21431174)×1
检测结果解释:

| 拷贝数变异 | 染色体区域 | 最小间隔 | 最短长度(Mb) | 探针数目 | 最大间隔 | 最长长度(Mb) |
|---|---|---|---|---|---|---|
| 缺失 | 22q11.21 | 18918741-21431174 | 2.512 | 857 | 18918473-21797384 | 2.879 |
以上异常区域包含超过20个基因。
此区域的缺失导致22q11.2缺失综合征(22q11.2 deletion syndrome,DiGeorge/Velocardiofacial syndrome,OMIM:#188400)。该疾病的临床症状包括发育迟缓与语言障碍,智力障碍,先天性心脏病,颚裂,低血钙,免疫缺陷等。
检验方法:
将患者的DNA和同性别的正常对照DNA杂交到XXXXXXXX芯片(芯片版本CMAXXXX,芯片编号XXXXXXXXXXXX-X),分析杂交信号,检测受检样本的染色体拷贝数异常情况。此芯片在临床疾病相关的染色体区域的平均分辨率为xx kb,可检测到的最小缺失和扩增为xx kb。数据分析和解析依据本实验室的内部数据库和公众数据库,包括UCSC Genome Browser,Database of Genomic Variants (DGV),Mutation Database (HGMD), DECIPHER以及科学文献。我们对于小于300 kb的无已知表型的拷贝数变化将酌情根据具体情况决定是否给予报告。
建议:
建议至临床医师进行就诊,结合其他临床数据和/或FISH结果进行明确诊断,并进行遗传咨询。建议对父母进行进一步FISH检测。
局限性:
此方法仅用于检测拷贝数异常导致的缺失和重复,无法检测到拷贝数正常的染色体变化,如平衡易位,倒位或平衡插入。此方法亦无法可靠地检测低于10%的染色体嵌合体,亦无法检测探针未覆盖的染色体区域。也不能用于DNA序列的变化,如点突变,小片段插入和缺失等的检测。
声明:
1.报告结果只对本次送检样本负责。
2.关于本报告的其他问题或需要进行相关健康咨询服务,请联系检测实验室。
电子邮箱:info@dnalaboratory.com 联系电话:(123) 456-7890
参考文献:https://www.ncbi.nlm.nih.gov/books/NBK1523/
报告:XX,医学博士,2018年3月20日上午10:41
审核:XX,医学博士,2018年3月20日下午12:30
电子签名:XX,医学博士,2018年3月20日下午12:30

肿瘤体细胞变异的染色体微阵列芯片检测报告
结果:阳性
检测样本:骨髓穿刺
检测结论:异常,检测到多处基因拷贝数异常,包括位于染色体9p21.3的CDKN2A基因缺失,位于染色体7p12.2处的IKZF1基因缺失,以及位于染色体Xp22.33处的缺失导致的P2RY8-CRLF2融合基因。以上发现在B细胞急性淋巴细胞白血病(B-ALL)患者中提示预后不良。
检测结果解释:
位于染色体9p21.3处的缺失包含CDKN2A基因在内的多个基因,与应用CDKN2A/p16探针进行荧光原位杂交(FISH)检测结果一致。含CDKN2A基因在内的部分区域为纯和缺失。位于染色体7p14.1p12.1处的缺失约13 Mb大小,包括IKZF1基因。位于染色体Xp22.33处的缺失导致P2RY8-CRLF2融合基因的形成。此分析检测到的多处基因拷贝数异常,在B细胞急性淋巴细胞白血病(B-ALL)患者中提示预后不良。
详细检测结果
arr[GRCh37] 4q31.21q35.2(143,702,364_190,957,473)×1[0.89] ~47 Mb,
6q24.1q24.2(140,838,649_143,869,867)×1[0.86] ~3 Mb,
7p14.1p12.1(38,535,159_51,586,184)×1[0.82] ~13 Mb,包括IKZF1
7p14.3(32,660,523_33,295,330)×1[0.82] ~635KB,
7p21.3p21.2(7,595,777_14,598,532)×1[0.89] ~7 Mb,ETV1
9p13.2(37,036,795_37,258,316)×1[0.74] ~222KB,
9p22.1p21.3(19,584,383_24,144,035)×1[0.91] ~4.5 Mb,包括CDKN2A缺失,经FISH确证
9p21.3(21,976,745_21,999,069)×0 ~22 kb,部分CDKN2A基因
Xp22.33(1,324,508_1,651,464)×1[0.71] ~327 kb,形成P2RY8-CRLF2融合基因
Xq21.31q28(89,606,325_155,270,560)×3[0.96] ~65.7 Mb
检测方法:
应用Oligo-SNP (oligonucleotide,single nucleotide polymorphism,Affymetrix CytoScan HD)对全基因组范围内的拷贝数变异进行检测。该芯片包括190万个拷贝数探针和750万个SNP探针,平均探针间距为1150个碱基对。本报告阈值设定为大于200kb的拷贝数扩增,大于50 kb的拷贝数缺失以及大于10 Mb的纯合性片段。小于这些阈值的已知致病的异常变化亦有可能被酌情报告。
建议:
该检测结果须结合患者的临床表现和其他检测结果,如常规染色体分析和/或FISH检测。建议至临床医师进行明确诊断,并进行相关治疗。
局限性:
1.在异常细胞数目较低的情况下,此检测可能产生假阴性结果。
2.此方法仅用于检测拷贝数异常导致的缺失和重复,无法检测到拷贝数正常的染色体变化,如平衡易位,倒位或平衡插入。此方法亦无法可靠地检测低于10%的染色体嵌合体,亦无法检测探针未覆盖的染色体区域。也不能用于DNA序列的变化,如点突变,小片段插入和缺失等的检测。
声明:
1.报告结果只对本次送检样本负责。
2.关于本报告的其他问题或需要进行相关健康咨询服务,请联系检测实验室。
电子邮箱:info@dnalaboratory.com 联系电话:(123) 456-7890
参考文献:
N Engl J Med. 2009;360(5):470-80; Blood. 2010;115(26):5312-21; Hematology Am Soc Hematol Educ Program. 2012;389-96)。
报告:XX,医学博士,2018年3月20日上午10:41
审核:XX,医学博士,2018年3月20日下午12:30
电子签名:XX,医学博士,2018年3月20日下午12:30

基因检测报告
检测项目:全基因组测序;双脱氧链终止法测序验证
受检者临床指征:身材矮小和下肢弯曲
结论:阳性
检测结果可解释受检者临床指征
检测结论概述
受检者X染色体的PHEX基因上检出一个半合致病变异。因受检者母亲未检出相同变异,且全基因组测序数据证实亲生母子关系,该变异确认为新发。PHEX致病变异与X连锁显性遗传低磷性佝偻病相关。该检测结果可解释受检者的身材矮小和下肢弯曲。

| 基因&转录本 | 变异位点 | 区域 | 合子状态 | 染色体位置 | 疾病或表型 | 遗传模式 | 来源 | 分类 |
|---|---|---|---|---|---|---|---|---|
| PHEXNM_000444.5 | c.1586+1G>A | 14号内含子共22外显子 | 半合 | ChrX:22196494(GRCh37/hg19) | 低磷性佝偻病 | X连锁显性 | 新发 | 致病 |
建议
该结果需要结合受检者的临床表现、家族史和种族背景等进行综合分析。请结合受检者血的生化指标例,如血清磷等,和骨骼影像学表现进行进一步判断。建议该受检者及其亲属接受遗传咨询。关于本报告的其他问题或了解当地遗传咨询服务,请联系DNA实验室。电邮:info@dnalaboratory.com或致电:(123) 456-7890。
致病变异的详细信息

| 基因&转录本 | 变异位点 | 区域 | 合子状态 | 染色体位置 | 疾病或表型 | 遗传模式 | 来源 | 分类 |
|---|---|---|---|---|---|---|---|---|
| DWPHEXNM_000444.5 | c.1586+1G>A | 14号内含子共22外显子 | 半合 | ChrX:22196494(GRCh37/hg19) | 低磷性佝偻病 | X连锁显性 | 新发 | 致病 |
变异解读:PHEX的c.1586+1G>A新发半合变异已于两例患低磷性佝偻病的男孩中有报道1,2,但尚未在大规模人群基因研究数据中有报道。该变异发生在外显子+/-1,2碱基的剪接位点,预测可导致剪接改变从而引起蛋白缺失或功能异常。PHEX功能丧失是X连锁显性遗传低磷性佝偻病的致病机制。总之,根据该变异对蛋白功能影响的预测,在数例男性患者确认为新发半合变异,在普通人群中未曾报道,与患者表型和遗传模式相符,判断该变异为造成X连锁显性低磷性佝偻病的致病变异。【依据的ACMG/AMP标准:PVS1、PS2、PM2、PP4】3
背景知识:低磷性佝偻病是一组异质性的疾病,可为遗传性或获得性。其特征是肾脏排磷增加,导致低磷血症和低磷性佝偻病4。不同基因的致病性变异可导致不同模式的遗传性低磷性佝偻病5。PHEX为X连锁显性遗传低磷性佝偻病的常见致病基因(OMIM 307800)1,6。PHEX编码含749个氨基酸的跨膜肽链内切酶,参与骨和牙本质矿化和肾磷酸盐重吸收。X连锁低磷性佝偻病的发病率约为十万分之1.9至5 6,7,外显率100%,但临床表现存在异质性4。
亲属风险:PHEX相关的低磷性佝偻病循X连锁显性遗传模式。本例受检者会将此致病变异遗传给他所有的女儿,但不会遗传给儿子。遗传了该致病变异的后代将患此病,但严重程度不一。本例受检者检出新发变异,其母亲血液样品无变异。但由于不排除母亲生殖细胞嵌合的可能性,其母下一胎患此病的风险高于常人。
检测方法
应用未经PCR扩增的全基因组测序(WGS)技术在HiSeqX10平台上进行批量化DNA测序分析。对原始数据进行分析,筛选与身材矮小和骨骼畸形相关的候选基因,检测可能的致病变异(详见附加报告)。所有候选基因编码区的测序深度在8X以上的覆盖率均>99%。受检者测序结果用Burrows-Wheeler Aligner (BWA)与人类参考序列(GRCh37)进行比对,使用基因组分析软件包(GATK)鉴定出短小的核苷酸变异。拷贝数变异和结构变异的鉴定方法参见已发表的方法8,9。筛选候选基因致病变异的条件:(1)根据基因组集合数据库(gnomAD,http://gnomad.broadinstitute.org/),在所有种群中变异的等位基因频率≤5%;(2)在有相关临床表现的患者中已报道过;(3)预测可能影响基因功能;(4)若符合隐性遗传模式时,纯合或复合杂合;或(5)新发变异。对经过上述筛选策略筛选出来的变异,再根据公共或商业数据库的信息(如ClinVar、gnomAD、HGMD、LSDBs和dbSNP)、已发表的文献、患者的临床特征和家族史信息、共分离原则、功能研究以及生信预测等进行综合评估10。报道的变异根据HGVS命名标准(http://varnomen.hgvs.org/)。每个变异的临床意义根据ACMG/AMP指南进行评估3。本报告仅汇报与受检者临床表现相关的变异。报告的变异均用其他方法再次验证,包括用双脱氧链终止法测序验证短小的核苷酸变异,用定量微滴数字PCR验证拷贝数变异,和用双脱氧链终止法对长程PCR产物测序验证结构变异断点。
局限性
本测试未对受检者基因组的每个碱基进行测序,也无法检出受检者的所有基因变异。本报告仅局限于报告已有证据表明与受检者临床指征相关的变异。对变异的解读局限于目前已知的信息。目前全基因组测序方法还不能完全可靠地检出重复扩展的动态变异、拷贝数变异及结构变异等。因此,由于信息缺乏或技术所限,并非所有与受检者临床表现相关的基因或区域都能被检出或解读。
声明
全基因组测序由临床基因测序实验室(XX省XX市;CLIA# 22D2055555)完成。生物信息学分析、变异位点验证和解释均由DNA实验室完成(XX省XX市;CLIA#22D2056666)。本检测由DNA实验室研发,质控标准经过本实验室核实。本检测尚未被联邦食品和药物管理局(FDA)批准或通过,但目前此类检测的临床应用不必经FDA批准。本检测结果不能作为临床诊断或受检者健康管理决策的唯一凭据。
参考文献
1.Capelli S, Donghi V, Maruca K, et al. Clinical and molecular heterogeneity in a large series of patients with hypophosphatemic rickets. Bone 2015;79:143-9.
2.Ran Q, Xiong F, Zhu M, et al. [Novel PHEX gene mutations in patients with X-linked hypophosphatemic rickets: an analysis of 2 cases]. Zhongguo Dang Dai Er Ke Za Zhi 2017;19:534-8.
3.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in medicine : official journal of the American College of Medical Genetics 2015;17:405-24.
4.Ruppe MD. X-Linked Hypophosphatemia. In: Adam MP, Ardinger HH, Pagon RA, et al., eds. GeneReviews((R)). Seattle (WA)1993.
5.Razali NN, Hwu TT, Thilakavathy K. Phosphate homeostasis and genetic mutations of familial hypophosphatemic rickets. Journal of pediatric endocrinology & metabolism : JPEM 2015;28:1009-17.
6.Rafaelsen S, Johansson S, Raeder H, Bjerknes R. Hereditary hypophosphatemia in Norway: a retrospective population-based study of genotypes, phenotypes, and treatment complications. European journal of endocrinology / European Federation of Endocrine Societies 2016;174:125-36.
7.Beck-Nielsen SS, Brock-Jacobsen B, Gram J, Brixen K, Jensen TK. Incidence and prevalence of nutritional and hereditary rickets in southern Denmark. European journal of endocrinology / European Federation of Endocrine Societies 2009;160:491-7.
8.Dong Z, Xie W, Chen H, et al. Copy-Number Variants Detection by Low-Pass Whole-Genome Sequencing. Current protocols in human genetics / editorial board, Jonathan L Haines [et al] 2017;94:8 17 1-8 6.
9.Dong Z, Wang H, Chen H, et al. Identification of balanced chromosomal rearrangements previously unknown among participants in the 1000 Genomes Project: implications for interpretation of structural variation in genomes and the future of clinical cytogenetics. Genetics in medicine : official journal of the American College of Medical Genetics 2017.
10.Duzkale H, Shen J, McLaughlin H, et al. A systematic approach to assessing the clinical significance of genetic variants. Clinical genetics 2013;84:453-63.
报告:XX,医学博士,2018年3月18日上午10:41
审核:XX,博士,FACMG,2018年3月18日下午12:30
电子签名:XX,博士,FACMG,2018年3月18日下午12:30

基因检测附加报告
检测项目:全基因组测序;双脱氧链终止法测序验证
受检者临床指征:身材矮小和下肢弯曲
分析的目标基因及其覆盖率
本测试重点分析了已报道过与身材矮小和下肢弯曲有关的149个基因,如下表所列。请注意,本检测不能完全排除致病变异在所列基因未完全覆盖的区域中或在本清单中未包含的基因中的可能。
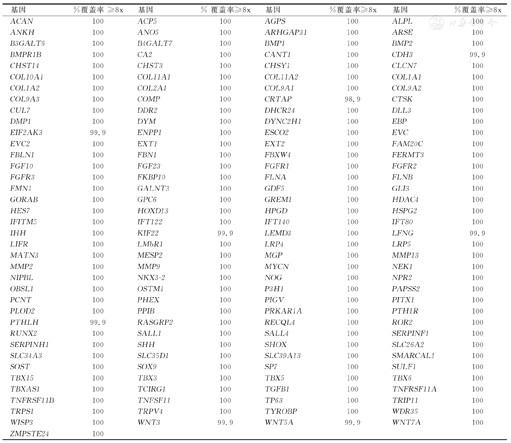
| 基因 | %覆盖率≥8x | 基因 | %覆盖率≥8x | 基因 | %覆盖率≥8x | 基因 | %覆盖率≥8x |
|---|---|---|---|---|---|---|---|
| ACAN | 100 | ACP5 | 100 | AGPS | 100 | ALPL | 100 |
| ANKH | 100 | ANO5 | 100 | ARHGAP31 | 100 | ARSE | 100 |
| B3GALT6 | 100 | B4GALT7 | 100 | BMP1 | 100 | BMP2 | 100 |
| BMPR1B | 100 | CA2 | 100 | CANT1 | 100 | CDH3 | 99.9 |
| CHST14 | 100 | CHST3 | 100 | CHSY1 | 100 | CLCN7 | 100 |
| COL10A1 | 100 | COL11A1 | 100 | COL11A2 | 100 | COL1A1 | 100 |
| COL1A2 | 100 | COL2A1 | 100 | COL9A1 | 100 | COL9A2 | 100 |
| COL9A3 | 100 | COMP | 100 | CRTAP | 98.9 | CTSK | 100 |
| CUL7 | 100 | DDR2 | 100 | DHCR24 | 100 | DLL3 | 100 |
| DMP1 | 100 | DYM | 100 | DYNC2H1 | 100 | EBP | 100 |
| EIF2AK3 | 99.9 | ENPP1 | 100 | ESCO2 | 100 | EVC | 100 |
| EVC2 | 100 | EXT1 | 100 | EXT2 | 100 | FAM20C | 100 |
| FBLN1 | 100 | FBN1 | 100 | FBXW4 | 100 | FERMT3 | 100 |
| FGF10 | 100 | FGF23 | 100 | FGFR1 | 100 | FGFR2 | 100 |
| FGFR3 | 100 | FKBP10 | 100 | FLNA | 100 | FLNB | 100 |
| FMN1 | 100 | GALNT3 | 100 | GDF5 | 100 | GLI3 | 100 |
| GORAB | 100 | GPC6 | 100 | GREM1 | 100 | HDAC4 | 100 |
| HES7 | 100 | HOXD13 | 100 | HPGD | 100 | HSPG2 | 100 |
| IFITM5 | 100 | IFT122 | 100 | IFT140 | 100 | IFT80 | 100 |
| IHH | 100 | KIF22 | 99.9 | LEMD3 | 100 | LFNG | 99.9 |
| LIFR | 100 | LMbR1 | 100 | LRP4 | 100 | LRP5 | 100 |
| MATN3 | 100 | MESP2 | 100 | MGP | 100 | MMP13 | 100 |
| MMP2 | 100 | MMP9 | 100 | MYCN | 100 | NEK1 | 100 |
| NIPBL | 100 | NKX3-2 | 100 | NOG | 100 | NPR2 | 100 |
| OBSL1 | 100 | OSTM1 | 100 | P3H1 | 100 | PAPSS2 | 100 |
| PCNT | 100 | PHEX | 100 | PIGV | 100 | PITX1 | 100 |
| PLOD2 | 100 | PPIB | 100 | PRKAR1A | 100 | PTH1R | 100 |
| PTHLH | 99.9 | RASGRP2 | 100 | RECQL4 | 100 | ROR2 | 100 |
| RUNX2 | 100 | SALL1 | 100 | SALL4 | 100 | SERPINF1 | 100 |
| SERPINH1 | 100 | SHH | 100 | SHOX | 100 | SLC26A2 | 100 |
| SLC34A3 | 100 | SLC35D1 | 100 | SLC39A13 | 100 | SMARCAL1 | 100 |
| SOST | 100 | SOX9 | 100 | SP7 | 100 | SULF1 | 100 |
| TBX15 | 100 | TBX3 | 100 | TBX5 | 100 | TBX6 | 100 |
| TBXAS1 | 100 | TCIRG1 | 100 | TGFB1 | 100 | TNFRSF11A | 100 |
| TNFRSF11B | 100 | TNFSF11 | 100 | TP63 | 100 | TRIP11 | 100 |
| TRPS1 | 100 | TRPV4 | 100 | TYROBP | 100 | WDR35 | 100 |
| WISP3 | 100 | WNT3 | 99.9 | WNT5A | 99.9 | WNT7A | 100 |
| ZMPSTE24 | 100 |

基因检测次要和意外发现报告
检测项目:全基因组测序;双脱氧链终止法测序验证
受检者临床指征:身材矮小和下肢弯曲
结论:无次要发现;意外发现致病基因携带者状态
检测结论概述
对受检者进行全基因组测序,未检出ACMG推荐检测基因(https://www.ncbi.nlm.nih.gov/clinvar/docs/acmg/ 1)的致病变异。但是,在本检测中意外发现受检者携带一个GJB2的杂合致病变异。该变异与常染色体隐性遗传的非综合征型耳聋DFNB1相关。本受检者的该变异遗传自其父亲。本发现与该受检者本次检测的临床指征无关。

| 基因&转录本 | 变异位点 | 区域 | 合子状态 | 染色体位置 | 疾病或表型 | 遗传模式 | 来源 | 分类 |
|---|---|---|---|---|---|---|---|---|
| GJB2 NM_004004.5 | c.235delCp.Leu79fs | 2号外显子共2个外显子 | 杂合 | Chr13:20763486(GRCh37/hg19) | 非综合征型耳聋 | 常染色体隐性 | 父亲 | 致病 |
建议
该结果需要结合受检者的临床表现、家族史和种族背景等进行综合分析。建议该受检者及其亲属接受遗传咨询。关于本报告的其他问题或了解当地遗传咨询服务,请联系DNA实验室。电邮:info@dnalaboratory.com或致电:(123) 456-7890。
变异的详细信息

| 基因&转录本 | 变异位点 | 区域 | 合子状态 | 染色体位置 | 疾病或表型 | 遗传模式 | 来源 | 分类 |
|---|---|---|---|---|---|---|---|---|
| GJB2 NM_004004.5 | c.235delCp.Leu79fs | 2号外显子共2个外显子 | 杂合 | Chr13:20763486(GRCh37/hg19) | 非综合征型耳聋 | 常染色体隐性 | 父亲 | 致病 |
变异解读:GJB2的c.235delC(p.Leu79fs)变异是非综合征型隐性耳聋的常见致病变异。已有大量文献报道该变异的纯合子或与其他DFNB1致病变异的复合杂合子患有耳聋并在多名亲属中与表型共分离2-4 。基因组集合数据库gnomAD(http://gnomad.broadinstitute.org/variant/13-20763485-AG-A)中,该变异在东亚人群中的等位基因频率为0.6%(121/18870),与普通人群中常染色体隐性遗传性非综合征性耳聋的携带者频率相符合。该变异可导致移码,从而改变编码蛋白质的第12个氨基酸以后的序列并导致下游3个氨基酸序列后提前终止。上述改变预测可影响GJB2的蛋白功能。总之,根据ACMG/AMP标准,该变异为导致隐性遗传性非综合征型耳聋的致病变异。【依据的ACMG/AMP标准:PM4_Strong、PS4、PM3_Strong】5
背景知识:先天性耳聋是最常见的感觉功能缺陷。每千个新生儿中有1-2例为先天性耳聋患者。在DFNB1基因座(OMIM#220290)上GJB2相关的非综合征型耳聋,以先天性非进行性常染色体隐性遗传、轻度至极重度感音神经性耳聋为特征6。编码连接蛋白26的GJB2双等位基因致病变异是最常见的常染色体隐性非综合征型耳聋的致病原因。GJB2致病基因变异的杂合携带者不患有GJB2相关性耳聋。
亲属风险:GJB2的c.235delC(p.Leu79fs)变异导致的耳聋是常染色体隐性遗传病。若父母双方均为GJB2致病变异的杂合携带者,则其后代有25%的概率患GJB2相关性耳聋。本受检者为GJB2致病变异的杂合携带者,有50%的概率将该变异遗传给其子女。
检测方法
应用未经PCR扩增的全基因组测序(WGS)技术在HiSeqX10平台上进行批量化测序分析。对原始数据进行分析,筛选ACMG推荐报告的可导致儿童期发病的基因,分析可能的致病变异1,以及可导致严重隐性遗传病携带者状态的致病变异。所有候选基因的测序深度在8X以上的覆盖率均>99%。受检者测序结果用Burrows-Wheeler Aligner (BWA)与人类参考序列(GRCh37)进行比对,使用基因组分析软件包(GATK)鉴定出短小的核苷酸变异。拷贝数变异和结构变异的鉴定方法参见已发表的方法7,8。筛选候选基因致病变异的条件:(1)已报道过的变异;(2)以丧失基因功能为已知致病机制的基因中预测为失去功能的变异(即满足ACMG/AMP: PVS1);或(3)父母亲子关系核实情况下确定的新发变异(即满足ACMG/AMP: PS2)5。对经过上述筛选策略筛选出来的变异进行表型相关的因果评估。对通过上述策略筛选出来的变异再根据公共或商业数据库的信息(如ClinVar、gnomAD、HGMD、LSDBs和dbSNP)、发表的文献、相关疾病的临床特征和家族史信息、共分离原则、功能研究以及计算机预测等进行综合评估9。报道的变异根据HGVS (http://varnomen.hgvs.org/)命名。根据ACMG/AMP指南进行评估每个变异的临床意义5。本报告仅报道ACMG推荐报告的导致儿童期发病相关基因的致病变异和导致严重隐性遗传病的致病变异的携带者状态。报告的变异均用其他方法再次验证,包括用双脱氧链终止法测序验证短小的核苷酸变异,用定量微滴数字PCR验证拷贝数变异,和用双脱氧链终止法对长程PCR产物测序验证结构变异断点。
局限性
本测试未对受检者基因组的每个碱基进行测序,也无法检出受检者的所有基因变异。本报告仅局限于ACMG推荐检测的且已有证据表明可导致儿童期发病以及严重隐性遗传病的携带者状态的致病变异。目前全基因组测序方法还不能完全可靠地检出重复扩展的动态变异、拷贝数变异及结构变异。因此,由于信息缺乏或技术所限,并非所有相关的致病变异都能被检出或解读。
声明
全基因组测序由临床基因测序实验室(XX省XX市;CLIA# 22D2055555)完成。生物信息学分析、变异位点验证和解释均由DNA实验室完成(XX省XX市;CLIA#22D2056666)。本检测由DNA实验室研发,质控标准经过本实验室核实。本检测尚未被联邦食品和药物管理局(FDA)批准或通过,但目前此类检测的临床应用不必经FDA批准。本检测结果不能作为临床诊断或受检者健康管理决策的唯一凭据。
参考文献
1.Kalia SS, Adelman K, Bale SJ, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genetics in medicine : official journal of the American College of Medical Genetics 2017;19:249-55.
2.Kudo T, Ikeda K, Kure S, et al. Novel mutations in the connexin 26 gene (GJB2) responsible for childhood deafness in the Japanese population. Am J Med Genet 2000;90:141-5.
3.Abe S, Usami S, Shinkawa H, Kelley PM, Kimberling WJ. Prevalent connexin 26 gene (GJB2) mutations in Japanese. Journal of medical genetics 2000;37:41-3.
4.Rabionet R, Gasparini P, Estivill X. Molecular genetics of hearing impairment due to mutations in gap junction genes encoding beta connexins. Human mutation 2000;16:190-202.
5.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in medicine : official journal of the American College of Medical Genetics 2015;17:405-24.
6.Smith RJH, Jones MKN. Nonsyndromic Hearing Loss and Deafness, DFNB1. In: Adam MP, Ardinger HH, Pagon RA, et al., eds. GeneReviews((R)). Seattle (WA)1993.
7.Dong Z, Wang H, Chen H, et al. Identification of balanced chromosomal rearrangements previously unknown among participants in the 1000 Genomes Project: implications for interpretation of structural variation in genomes and the future of clinical cytogenetics. Genetics in medicine : official journal of the American College of Medical Genetics 2017.
8.Dong Z, Xie W, Chen H, et al. Copy-Number Variants Detection by Low-Pass Whole-Genome Sequencing. Current protocols in human genetics / editorial board, Jonathan L Haines [et al] 2017;94:8 17 1-8 6.
9.Duzkale H, Shen J, McLaughlin H, et al. A systematic approach to assessing the clinical significance of genetic variants. Clinical genetics 2013;84:453-63.
报告:XX,医学博士,2018年3月19日下午4:15
审核:XX,博士,FACMG,2018年3月20日下午4:38
电子签名:XX,博士,FACMG,2018年3月20日下午4:38

























