
版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
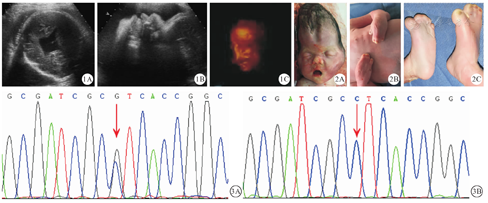
34岁,孕二产一,丈夫39岁,育有一子,现4岁,体健。孕妇平素月经规律,夫妇均无先天性疾病及家族遗传病史,无烟酒不良嗜好,否认孕期病毒感染、毒物和放射线接触史,无水源污染史。孕期未定期行产检。孕12周超声检查胎儿颈项透明层厚度(nuchal translucency,NT)为1.0 mm,孕18周血清学筛查为低风险,孕24周在外院进行系统超声检查未见明显异常。孕34周+3时在我院进行常规产前检查,超声显示胎儿双顶径测不出(因头型不规则),头围30.6 cm,腹围33.5 cm,股骨长径6.6 cm,胎心搏动规律,羊水指数14 cm,胎盘位于前壁,成熟度Ⅱ级;胎儿头型不规则,鼻根凹陷,前额突出明显,丘脑融合,双侧脑室体部融合(图1),手指、足趾因孕周及体位因素显示不清。超声检查提示胎儿为全前脑畸形,疑诊为Apert综合征。孕妇坚决要求引产。经我院伦理委员会讨论同意后终止妊娠。于34周+6引产,娩出一死男婴,可见特殊面容、并指(趾)(图2)。在征得其亲代知情同意后,留取引产胎儿皮肤组织,提取DNA并进行基因检测,同时抽取其父母外周血样进行验证。基因检测结果显示胎儿FGFR2基因的第7外显子中存在c.758C>G(P253R)杂合突变,其父母均未携带该突变(图3)。综合其临床表现和基因检测结果,将胎儿诊断为Apert综合征。
Apert综合征(OMIM 101200)是一种罕见的多颅缝早闭综合征,发病率为1/65 000[1]。本病主要表现为颅缝早闭所致的头颅异常、突眼和面中部严重发育不良、手/足并指(趾)畸形。目前,产前超声诊断的Apert综合征的报道较少。
Apert综合征是一种罕见的常染色体显性遗传病[2],多为散发病例,而患者的后代有50%的遗传风险。有研究显示,99%的Apert综合征是由于成纤维细胞生长因子受体2(FGFR2)基因S252W和P253R错义突变所致[3]。FGFR属于受体型蛋白酪氨酸激酶,成纤维细胞生长因子(FGF)与FGFR结合形成的信号通路在胚胎发育、骨骼形成、纤维化等方面发挥着重要的作用。间充质干细胞和成骨细胞的增殖和分化是重要的骨形成过程。FGFR2基因的S252W和P253R均为功能增强型突变。有研究表明,S252W突变可通过增强Runx2的表达,从而促进成骨细胞分化,最终导致颅缝早闭。也有研究者[4]报道,FGFR2 S252W功能获得性突变可导致细胞增殖增加和未成熟成骨细胞分化减少。相反,Chen等[5]通过FGFR2 S252W小鼠模型,证实增强的FGFR2活性可能诱导骨细胞凋亡的增加,从而导致冠状缝早闭。Luo等[6]用FGFR2 P253R小鼠模型模拟人类Apert综合征,通过在成体骨、软骨和中枢神经系统祖细胞中分别对FGFR2进行P253R突变,发现异常的颅骨形态是由颅底,脑底部和脑组织发育不良的综合作用引起的。这些发现加深了我们对于Apert综合征异常颅骨形态发病机制的认识,为进一步分析该病的颅骨表型以及临床治疗提供了新的线索。
本例胎儿NT无增厚,中孕期唐氏综合征筛查为低风险,故未做羊水染色体分析。有研究表明,46.9%的Apert综合征患儿的父亲年龄≥ 35岁[7]。本例胎儿父亲39岁,与文献报道相符。本研究中,患儿的基因突变为P253R,表现为颅面部骨骼畸形和手足并指(趾)畸形,但父母表型正常,且均未检测到上述突变,提示患儿为新发突变。
Apert综合征的产前超声图像特征为前额隆起、尖颅、短头,头颅形状不规则,双侧肢体对称性并指(趾),颜面部正中矢状切面轮廓线异常,面容特殊合并其他畸形[8]。产前超声显示胎儿头型不规则、特殊面容以及合并侧脑室融合,引产后发现胎儿指(趾)融合,并经过基因诊断进一步确诊为Apert综合征。
Apert综合征由于颅面骨的畸形和指(趾)融合,患儿出生后多死于呼吸道梗阻和颅内压增高引起的并发症,后期可出现视力障碍和智力低下等,需进行多次手术治疗[9],给家庭带来沉重的经济和心理负担。本例胎儿侧脑室前角和后角发育正常,外院孕中期超声检查未见异常,但随着孕周的增加,侧脑室体部融合更加明显,表现为特殊面容,至孕晚期超声检查时才发现。因此,对于Apert综合征进行定期产前检查非常重要。Apert综合征不仅有颅面部的特殊畸形,还可能合并多种手足畸形。产前超声可根据胎儿颅面部的典型表现及早发现Apert综合征,对孕妇决定是否继续妊娠、新生儿的早期治疗以及再次妊娠时的遗传咨询均有重要的参考价值。
所有作者均声明不存在利益冲突

























