
版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
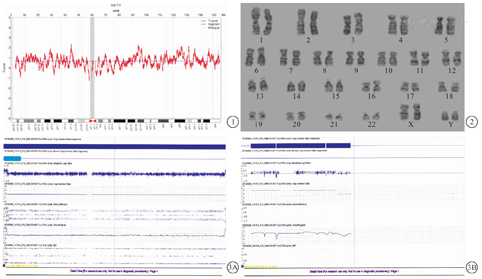
29岁,汉族,G3P1,孕16+2周入院产检,借助政府惠民政策在我院行无创产前检测(non-invasive prenatal testing,NIPT),提示X染色体数目增多(图1)。患者既往体健,月经规律,人工流产及剖宫产各1次,否认其他手术史,否认心、肺、脑等器官的基础疾病,否认遗传病家族史,孕期无有毒有害物质接触史和用药史,无药物和食物过敏史。丈夫31岁,夫妻双方表型正常,非近亲婚配。女儿6岁,表型正常。孕21+2周B超检查未见异常。在征得其知情同意后,在B超引导下行羊膜腔穿刺,抽取羊水25 mL,进行染色体核型分析和染色体微阵列分析。细胞遗传学检查:常规培养羊水细胞,制备染色体,G显带,用莱卡GSL-120全自动染色体核型扫描仪扫描核型,镜下计数30个核型,分析7个,结果为48,XXYY(图2),Affymetrix CytoScan 750K SNP-Array微阵列检测显示胎儿X和Y染色体均为二倍体,证实其为48,XXYY(图3)。孕妇于26+6周选择终止妊娠。夫妻双方外周血染色体核型分析均未见异常。
48,XXYY综合征是一种罕见的性染色体数目异常,在男性新生儿中的发病率为1/40 000~1/18 000[1]。多余的X染色体可导致睾丸发育不全和性腺功能低下,与克氏综合征(47,XXY)相近,因此曾被认为是克氏综合征的一个变种。近年来,研究者发现48,XXYY综合征患者在外貌特征、精神行为特点上仍与克氏综合征存在差异。
48,XXYY综合征的常见临床表现包括小睾丸、隐睾、性腺功能减退、不育、智力低下、青光眼和肘无法外翻等[2]。患者在胎儿期和婴儿期多无异常表现或仅有轻微表型,借助超声和血清学检查难以发现,多数患者在青春期后被发现。已发现的病例大多为成年患者,在产前发现者非常少见。48,XXYY综合征性染色体异常形成的原因是亲代一方或双方的生殖细胞在减数分裂过程中发生了性染色体不分离。
NIPT作为产前筛查染色体非整倍体的一种重要手段,可通过测定胎儿X和Y染色体的数目评估胎儿罹患性染色体非整倍体的风险。但Y染色体上的重复序列增加了准确比对的难度,而无创检测胎儿X染色体的数目也受到以下因素的影响:(1)母体或胎盘存在X染色体非整倍体嵌合;(2)高龄孕妇发生X染色体丢失[3]。鉴于上述原因,NIPT技术无法准确区分携带性染色体非整倍体男性胎儿X、Y染色体的拷贝数,需借助羊膜腔穿刺进行确诊。本例胎儿NIPT X染色体Z值=14.308(正常参考值范围:-3~+3),X、Y染色体的浓度分别为2.643%和9.753%,经生物信息学分析提示胎儿性染色体数目增多。对于NIPT提示胎儿性染色体非整倍体高风险的孕妇,应告知其检测的局限性,建议进一步的产前诊断。
所有作者均声明不存在利益冲突

























