
版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
患儿 女,7岁4个月,确诊癫痫3年余,表现为无热抽搐、全身强直发作。体格检查:身高110 cm,体重15.3 kg(均相当于第3百分位),头围49.5 cm。体型消瘦,神志清楚,外观无明显畸形,四肢肌力及肌张力正常。家系调查:患儿有一弟,5月龄,表型正常。母亲无不良孕产史。父母体健,系非近亲结婚,否认抽搐、智力低下以及遗传代谢病家族史。患儿目前每天抽搐多次,均表现为意识模糊、双眼凝视、口唇发绀、全身强直发作。除首次抽搐时伴有38℃发热外,其余均不伴发热。
实验室检查:脑电图示左侧中颞区不规则尖波、尖慢波,可波及同侧前后颞区。脑电图检测中患儿抽搐1次,同期脑电图见广泛高电位尖慢、棘慢、多棘慢综合波发放。头颅磁共振成像提示髓鞘化延迟。垂体高度约4 mm。尿气相色谱-质谱筛查未见明显异常。智力量表测量提示为正常边缘水平。
细胞遗传学检查:在征得其父母知情同意后采集血样,常规进行外周血淋巴细胞培养,染色体制备,G显带分析,患儿核型为46,X,Xq+(图1)。其父母核型均未见异常。高通量测序未发现患儿携带相关致病的微小变异。基于二代测序进行拷贝数变异分析(copy number variantion sequencing, CNV-seq)提示患儿Xq22.1q23区存在约15.30 Mb的重复([hg19]99 551 265-115 594 027×3)(图2)。检索DECIPHER数据库,其重复片段所包含的基因如图3。本研究经天津市儿童医院伦理学委员会批准(2016021)。
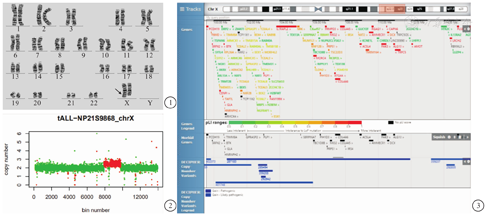
Xq22.1-Xq23重复既往报道很少,其范围涵盖PLP1、DCX、PAK3、PCDH19等功能基因。PCDH19的变异可导致癫痫性脑病9型,其特征为婴儿期癫痫发作、智力障碍、发育迟缓。首次发作年龄平均为14个月。该病仅影响杂合子女性[1]。有报道携带PCDH19变异的女性有自闭症的表现[2],且部分患者智力处于正常边缘水平[3]。Hirabayashi等[4]曾报道1例携带Xq21.32-q22.1区6.69 Mb缺失的患者,其缺失区涵盖PCDH19基因,表现为全身强直性癫痫发作。本例患儿的重复片段亦涵盖PCDH19基因,但文献中尚无类似重复致病的报道。患儿起病相对较晚,且无自闭症表现,其机制尚待进一步的研究。
PLP1基因与佩梅病(Pelizaeus-Merzbacher disease,PMD)相关。该病常见于男性,女性多为携带者。本例患儿的表现与PMD不符,亦不同于DCX、PAK3等其他功能基因变异所导致的表型。DECIPHER数据库收录有3例与本例患儿相似的重复片段,但其表型不尽相同。
相比于传统的染色体核型分析,CNV-seq分析可以更精确地定位大片段的缺失或重复,明确所涉及的关键基因,有利于快速、高效的诊断。
所有作者均声明不存在利益冲突

























