
版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
患儿 女,3岁1个月,系第一胎,孕37+3周因羊水少经剖宫产出生。出生时体重1.75 kg,头围40 cm。生后发现体格发育落后,体重增长不良,伴先天性心脏病(房间隔缺损)、右手拇指并指及左足第四脚趾畸形(图1A、图1B)。8月余能竖头,1岁余能独坐,于2岁8个月时来我院行"房间隔缺损修补术"。现可扶站,不能独站。查体:身高68 cm,体重6 kg,面部平坦、眼距宽、眼裂小、人中低平、鼻梁低平、球状鼻等特殊面容,低耳位,左手通贯掌,右手拇指并指,左足第四脚趾畸形。肌张力偏低,双侧巴氏征可疑阳性。
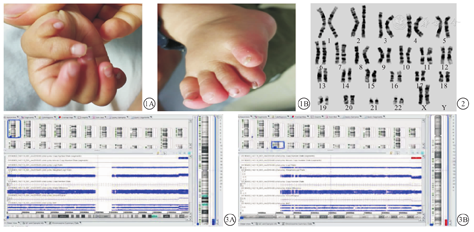
家系调查:父亲31岁,母亲28岁,否认近亲婚配及遗传病家族史。外周血染色体G显带核型分析结果:患儿母亲染色体核型为46,XX,t(1;15)(q42;q26),患儿及父亲染色体未见异常(图2)。经单核苷酸多态性微阵列芯片(single nucleotide polymorphism array, SNP-array)检测,患儿为arr[hg19]1q42.3q44(235 763 198-249 224 684)×3;arr[hg19]15q26.2q26.3(96 872 281-102 429 112)×1,即1号染色体q42.3q44区段有约13.4 Mb的重复;15号染色体q26.2q26.3区段有约4.6 Mb的缺失,见图3A、图3B。
本研究经郑州儿童医院伦理委员会批准(2020-K-23),患儿监护人签署知情同意书。
染色体微缺失微重复综合征(microdeletion and microduplication syndromes,MMS)是儿童先天性畸形及智力障碍的主要原因。在原因未明的发育迟缓、智力低下、自闭症及多发先天性畸形患者中,12%存在有临床意义的染色体微缺失微重复[1]。目前全世界已报道近300种MMS,大部分MMS以新发变异为主(约85%~95%),少部分为亲代遗传(约5%~10%)[2]。
本例患儿经SNP-array检出1号染色体q42.3q44区段重复约13.4 Mb,该重复片段在DECIPHER数据库有多例智力障碍、发育迟缓等病例报道。1q42-q44区域包含多个基因,已明确的与智力障碍有关的基因有ZBTBl8和FMN2。文献报道1q42q44重复的临床表型主要包括发育迟缓、智力障碍、大头畸形、三角脸、前额突出、鼻梁宽、人中异常、心脏缺陷等[3]。患儿另检出15号染色体长臂q26.2q26.3区段缺失约4.6 Mb,该区域包含15q26-qter缺失综合征(OMIM:612626)和单倍剂量不足基因IGF1R(OMIM:147370),该综合征的主要临床表型为发育迟缓、小头畸形、小颌畸形、先天性心脏病等[4]。本例患儿尚存在手足指(趾)畸形,与上述表型存在差异,一方面可能由于其携带的1q重复及15q缺失片段不一致,另一方面可能同时存在两段染色体变异。此病例同时伴1q重复及15q缺失实属罕见,未查询到文献报道。
染色体核型结果提示患儿母亲为t(1;15)(q42;q26)平衡易位携带者,而该易位正是患儿1q42.3q44区段微重复及15q26.2q26.3区段微缺失产生的原因。平衡易位携带者产生正常配子的概率极低,获得表型正常后代的理论概率仅为2/18,实际约为1/3[5]。本例患者形成基因拷贝数变异的机制可能由于亲代易位染色体断裂位点的不稳定性造成的,因染色体变异片段较小,因此未导致早期流产及胎死宫内的情况。
所有作者均声明不存在利益冲突

























