
版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
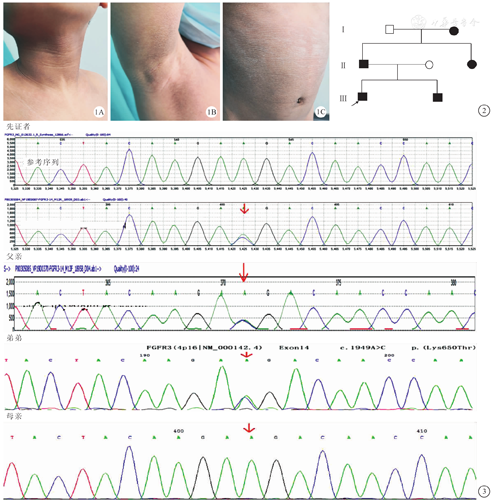
先证者 男,8岁5月龄,因"身材矮小2年余"就诊。患儿系第一胎,孕39周顺产,产时无窒息,出生时体重2.8 kg,身长51 cm。患儿出生后数月发现颈部、腋下肤色较深并逐渐加重,4岁左右腹部皮肤颜色加深并粗糙(未予诊治)。于6岁左右发现身高较同年人矮小,年生长速度不详。无其他不适,智力及精神运动发育均正常。无特殊疾病史。母亲孕期无异常,家族中无遗传病史。查体:身高118.7 cm(-2.5 SD),坐高64.1 cm,指间距117.0 cm,体重21.6 kg(-1.9 SD),体质指数(body mass index,BMI) 15.3 kg/m2 (P25-50th),头围51 cm。身材匀称,颈部、腋下、腹部见黑棘皮征(肤色呈褐色,粗糙,无脱屑)(图1)。心肺检查无异常,肝脾肋下未触及。会阴部无色素沉着,双侧睾丸2 mL,阴茎3.2 cm×1.5 cm,阴毛PH1期。
临床辅助检查:三大常规、肝肾功能、血脂、电解质、骨代谢指标均正常,甲状旁腺素、碱性磷酸酶、血清骨型碱性磷酸酶质量、25-羟基维生素D、空腹血糖、空腹胰岛素、空腹血清C肽、糖化血红蛋白、8AM皮质醇及血浆促肾上腺皮质激素均正常,类胰岛素样生长因子1(IGF-1)193 ng/mL(-1 SD),生长激素激发试验:生长激素峰值(60 min)7.83 μg/L,垂体MRI平扫+增强:未见明显异常。尺桡骨、胫腓骨、脊柱、骨盆、颅骨X线摄片等均未见异常。
家系调查(图2):先证者父母非近亲婚配,先证者父亲38岁,身高150 cm,其颈部、腋下、躯干可见黑棘皮征(其程度明显轻于先证者)。先证者母亲,36岁,身高158 cm,其皮肤未见黑棘皮征;遗传靶身高160 cm。先证者弟弟,4岁,身高95.6 cm(-2SD),颈部及腋下可见黑棘皮征(较先证者轻),腹部肤色正常;追问病史患儿姑姑(30岁,身高148 cm,未婚)、奶奶(身高145 cm)皮肤均有不同程度的黑棘皮征。
基因测序:经患儿父母知情同意并签署书面知情同意书,深圳市儿童医院伦理委员会审查批准( 20180302),采集患儿及其父母、弟弟外周血样各4 mL,标本委托广州金域医学检验中心进行医学全外显子基因检测。阳性基因变异类型进一步采用Sanger测序验证。患儿及其父亲、弟弟FGFR3基因的第14外显子上均存在c.1949A>C杂合错义变异,使得其所编码蛋白FGFR3的第650位氨基酸由赖氨酸变异为苏氨酸(p.Lys650Thr),其母亲的第14外显子未发生变异(图3)。患儿奶奶及姑姑未接受基因检查。
生物信息学及致病性分析:在千人基因组数据库(1000 Genome)、人类基因突变数据库HGMD、ESP6500和dbSNP数据库中查询变异情况;根据2015年美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics, ACMG)指南进行致病性分析[1]:(1)患儿、患儿父亲及患儿弟弟均检测出同一位点的相同变异,且三人具有相同的临床表现(支持,PP1);(2)该变异位于TD2结构域中激酶活性环区内,为热点变异。具有高度保守性(中等,PM1);(3)dbSNP数据库有收录(rs 121913105);另有变异p.(Lys650Asn)、p.(Lys650Gln)、p.(Lys650Met)均被HGMD数据库收录为致病性变异(强,PS1);(4)有文献报道在家族性黑棘皮病并身材矮小患者中检测到该变异(中等,PM5);(5)该突变为错义变异,错义变异所在的基因存在极少数的良性变异,极大多数是致病变异(支持,PP2);(6)生物信息软件(SIFT和PolyPhen-2)预测均有害,且变异位点在不同物种间高度保守(支持,PP3)。综上所述,该变异存在1个强、2个中等、3个支持证据,故认为该变异为致病性变异。
治疗:给予先证者rhGH(粉剂,安徽安科生物工程股份有限公司生产,生产批号为20170318)0.15 U/(kg·d)每晚睡前皮下注射,治疗2年。身高由治疗前118.7 cm(-2.5 SD)增长至133.9 cm(-1.4 SD),2年共增长15.2 cm。定期复查空腹血糖、空腹胰岛素、空腹血清C肽、糖化血红蛋白均在正常参考范围,类胰岛素样生长因子1(IGF-1)556 ng/mL(+1.5 SD)。皮肤肉眼观黑棘皮征较2年前有所加重。
黑棘皮病(acanthosis nigricans, AN)是一种非炎症性皮肤病,以皮肤增厚、粗糙、深褐色、具有天鹅绒般的质地为特征,主要见于皮肤皱褶处如颈部、腋下。黑棘皮病的发病机制大多数理论认为与胰岛素生长因子、酪氨酸激酶受体、表皮生长因子受体(EGFR)和成纤维细胞生长因子受体(fibroblast growth factor receptor, FGFR)有关,它们通过不同的信号通路刺激角质形成细胞和成纤维细胞的生成和增殖。家族性黑棘皮病为常染色体显性遗传,极少数报道为自发性突变;FGFR3基因变异已被确定与家族性AN有关,且常导致骨骼发育异常[2,3,4,5,6]。
人 FGFR3基因定位于染色体4p16.3区,cDNA长4.4 kb,共包含19个外显子,编码的FGFR3受体蛋白含有840个氨基酸残基,FGFR3蛋白全长由胞外区、单个疏水跨膜段和胞浆酪氨酸激酶结构域组成,蛋白的胞外部分与成纤维细胞因子结合,启动一系列下游信号STAT1和MEK/MAPK通路导致FGFR3信号的结构性激活,影响有丝分裂和分化,最终在骨的发育和维持中发挥作用,该基因在成员间高度保守,是长骨生长的抑制因子[2,7,8],该基因激活性变异可导致多种类型的骨骼发育不良[7],我们之前报道了FGFR3基因第10外显子发生c.1138G>A杂合变异导致的家族性软骨发育不全病例[9]。
位于FGFR3酪氨酸激酶结构域的第650位密码子(p.Lys 650)是FGFR3生物功能的重要残基,该位置发现了多种变异,其不同的碱基变异类型导致的骨骼异常的程度不同[10,11],如软骨发育不全中发现p. K650N和p.K650Q变异;致死性软骨发育不全中发现p.K650E变异;严重软骨发育不全伴发育迟缓并黑棘皮征中发现p.K650M变异;而p.K650T变异在软骨发育不全并黑棘皮征中发现,有些家族仅仅表现为黑棘皮征而无骨骼发育异常。p.K650T变异导致家族性黑棘皮征在国内尚未见报道,除身材矮小外无骨骼发育异常,与有关报道相同[2]。虽已明确FGFR3基因p.K650T变异导致黑棘皮征,但不能明确p.K650T变异对骨骼发育没有影响。Bellus等[11,12]认为FGFR3基因第650密码子变异是通过稳定激活状态构象而导致酪氨酸激酶结构域的结构性激活,其激活程度与该位点氨基酸残基的不同而变化,激活程度与变异引起的疾病严重程度成正比。与其他变异相比,p.K650T变异相对较温和,因此可无骨骼异常或骨骼改变轻微,但p.K650T变异导致角质形成细胞的生长增强和黑色素合成增加,可导致黑棘皮征。Yasuda等[13]报道由FGFR3基因变异引起的家族性AN其皮肤病变更广泛,本研究的家族成员中除先证者弟弟年幼,仅颈部出现黑棘皮征外,其他家族成员除颈部、腋下等皮肤皱褶处出现黑棘皮征,腹部均有不同程度的黑棘皮征。但患儿父亲黑棘皮征程度较轻。
FGFR3基因p.K650T变异,虽然部分患儿无骨骼发育异常,但存在身材偏矮或矮小;该家系受累者身材均矮小,先证者应用rhGH治疗2年,对其身高具有明显的改善,身高从治疗前的-2.5 SD增长到目前的-1.4 SD,但其黑棘皮征的程度较前有所加重。生长激素治疗可导致一过性的血糖及胰岛素升高,高胰岛素血症可导致黑棘皮征[14,15];另外,生长激素治疗可刺激类胰岛素生长因子1(IGF-1)的产生,IGF-1受体的激活可以导致许多不同细胞系的生长和分化,包括引起发育的角质形成细胞,从而导致黑棘皮征[15]。但到目前为止未见重组人生长激素治疗生长激素缺乏症患者出现黑棘皮征的报道;有关生长激素治疗软骨发育不全患儿是否导致黑棘皮征的报道结论不一[16,17]。该患儿治疗前后空腹血糖、空腹胰岛素、空腹C肽及胰岛素抵抗指数均在正常范围,没有明显变化,虽然治疗后IGF-1的水平有所升高,但仍在年龄别范围之内;患儿父亲、奶奶及姑姑均无肥胖及糖尿病病史,是否存在高胰岛素血症或胰岛素抵抗情况不详。因此,该患儿黑棘皮征程度加重是否与生长激素应用有关抑或与其自然病程有关尚需进一步研究。关于家族性黑棘皮征的治疗目前没有特效药物,有报道应用乙醇酸可改善黑棘皮征患者病变皮肤色素沉着及粗糙,且不良反应较轻[18]。
所有作者均声明不存在利益冲突

























