
版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
患儿 男,1岁4个月,运动及精神发育均落后,不能独站、行,仅能发单音节。患儿系足月顺产,出生体重3.70 kg,哭声弱,否认窒息缺氧史。其母届时32岁,父亲29岁。患儿有两个姐姐,生长发育均正常。患儿3月龄时曾因"室间隔缺损、房间隔缺损、动脉导管未闭、肺动脉高压"于外院手术治疗。
入院查体:体重8.3 kg(<第1百分位),身高76 cm(第3~ 10百分位),神清,精神可,反应一般,可辨亲疏,可逗笑,仅能发单音节,全身未见色素脱失斑,头型对称,头围42.5 cm,眼距稍宽,双侧外斜视,追物欠灵活(图1),听力粗检尚可,颈软,心肺腹部查体无明显阳性体征,阴茎短小,双侧阴囊空虚(图2),脊柱及四肢关节无畸形,四肢肌力Ⅳ级,肌张力低,双侧膝腱反射(++),双巴氏征(-),降落伞反射可引出。可肘支撑,不能直臂支撑,坐位平衡反射可引出,可短暂扶站,不能独站、行。
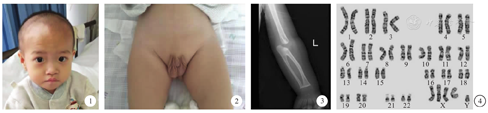
实验室检查:遗传代谢病筛查未发现特异性改变。心电图提示窦性心律、不完全性右束支传导阻滞。心脏彩超:无明显残余分流。腹部彩超:脾脏囊肿,中极0.8 cm × 0.7 cm。泌尿系彩超、髋关节平片、脑电图等均未见异常。X线检查提示左侧尺桡骨近端骨性融合,肘关节间隙增宽(图3)。脑干听觉诱发电位:两侧V波波形分化差,波幅低平。头颅磁共振平扫:脑实质内未见异常信号,侧脑室饱满。格里菲斯心理发育评估:粗大运动8个月,个人社会7.5个月,听力语言8个月,手眼协调10.5个月,视觉表现7.7个月,各领域百分位数均<1%,发育商50。染色体核型分析为49,XXXXY(图4)。
Klinefelter综合征又称先天性睾丸发育不全,最常见的核型为47,XXY,其余则主要包括额外的X或Y染色体或嵌合体。该病在男婴中的发生率为1/100 000~1/85 000,是男性不育最常见的遗传学病因。
49,XXXXY是Klinefelter综合征的一种罕见核型,其发生可能缘于初级或次级卵母细胞在减数分裂时的染色体不分离,从而形成包含4条X染色体的卵细胞与正常精子受精而成。Villamar等[1]借助X连锁限制性片段长度多态性分析,证实这类病例中大约88%的X染色体为母源性。
Peet等[2]通过对3个新病例的研究及对既往报道的100多例患者进行回顾,发现49,XXXXY综合征与Klinefelter综合征的典型核型47,XXY有不同的特征,提出了7种主要临床表现,包括特殊面容、特殊体形、多发性骨发育异常、生殖器缺陷、智力低下、语言障碍、先天性心脏病等,其中最重要的特征为先天性心脏病,最常见的疾病是动脉导管未闭。
国内在过去20年中曾报道8例49,XXXXY综合征,年龄最小者为1天,最大者为18岁,均存在生殖器异常,表现为阴茎短小、小睾丸或睾丸未降。有5例对患者的面容进行了描述,其共同表现包括眼距过宽、鼻根低平。5例存在先天性心脏病,包括房间隔缺损、室间隔缺损、动脉导管未闭、三尖瓣反流等。有2例具有骨发育异常,其中一例为典型的尺桡骨先天性融合。4例有发育迟缓、认知落后的表现[3]。肖华等[4]曾报告1例4月龄的患儿,外生殖器大小与同龄儿相仿,但外观明显异常,呈男性尿道下裂会阴型样表现,但尿道口位于阴茎头部。
本例患儿存在头围小、特殊面容、骨发育异常、生殖器缺陷、先天性心脏病、言语及运动发育落后等典型临床表现。由于其父母拒绝对其进行进一步的检查,无法排除是否同时存在其他基因组区域的异常。
Klinefelter综合征可伴发多种代谢异常,包括糖尿病、脂肪肝、高胆固醇血症、高尿酸血症等。国外报道合并发生糖尿病者多达15%~50%[5]。金玺等[6]曾报道1例18岁的49,XXXXY综合征患者,因"口干、多饮、多尿半月"就诊,入院查体发现性腺发育不全,追朔病史发现患者幼时即有运动及认知发育迟缓。
对49,XXXXY综合征患者应强调全面治疗,从11岁起用睾酮促进第二性征发育,同时强化教育与训练,以促进智力发育及正常性格的形成。
所有作者均声明不存在利益冲突

























