
女,1岁,系G2P2,足月顺产,2022年8月2日就诊于河南省儿童医院郑州儿童医院神经遗传代谢病科。出生体质量为3.35 kg,无窒息抢救史。患儿自3月龄至今患泌尿道感染4次、肺炎1次;4月余认生,现会发"ma ma"音;大运动发育稍落后,7个月会独坐,10个月会爬,现可扶站。出生4个月时血常规检查结果提示白细胞17.95 × 109/L(正常参考值为5.10 ~ 14.10 × 109/L),红细胞3.98 × 1012/L(正常参考值为4.00 ~ 5.50×1012/L),血红蛋白115 g/L(正常参考值为107 ~ 141 g/L),中性粒细胞占比68.2%(正常参考值为13.0% ~ 55.0%),淋巴细胞占比25.4%(正常参考值为33.0% ~ 77.0%),中性粒细胞12.23 × 109/L(正常参考值为0.80 ~ 5.80 × 109/L),淋巴细胞4.56 × 109/L(正常参考值为2.40 ~ 8.70 × 109/L),C-反应蛋白31.52 mg/L(正常参考值为0 ~ 10.00 mg/L)。尿常规检查结果提示红细胞25/高倍视野[正常参考值为(0 ~ 3)/高倍视野],白细胞146/高倍视野[正常参考值为(0 ~ 5)/高倍视野],诊断为泌尿道感染。膀胱输尿管造影检查提示双侧膀胱输尿管反流(图1),肾盂积水(分离10.8 mm,轻度)。心脏彩色多普勒超声检查提示心脏肌部室间隔缺损(图2A)、卵圆孔未闭(图2B)、左肺动脉中远端稍窄(图2C)。出生6个月头颅及腰骶部MRI检查提示白质髓鞘化落后,双侧侧脑室增宽(图3),左肾略大,肾盂周围异常信号,尾椎上翘,余腰骶椎MRI平扫未见明显异常。患儿父母表型未见异常,否认近亲结婚及家族遗传病史。患儿有1个6岁的哥哥,现体健。患儿入院时体质量为8 kg、身高为72 cm,头围44.5 cm,生长发育落后于同龄儿。精神可,有额头突出、眼窝凹陷、宽眼距、眼裂下斜、耳后旋、薄上唇、腰骶部隐窝等外观特征。肺、腹检查未见异常,四肢肌张力低下,双侧膝反射可引出。抽取患儿及其父母的外周血样,进行全外显子组测序分析,结果提示患儿染色体17p13.3区存在1.49 Mb微缺失:seq[GRCH37]del(17)(p.13.3p.13.3)chr7:g.882 518-2 371 192 del(图4),父母该区域均未发现相同的缺失。本研究通过了河南省儿童医院郑州儿童医院伦理委员会的审查(2022-K-070),患儿父母均签署了知情同意书。
版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
女,1岁,系G2P2,足月顺产,2022年8月2日就诊于河南省儿童医院郑州儿童医院神经遗传代谢病科。出生体质量为3.35 kg,无窒息抢救史。患儿自3月龄至今患泌尿道感染4次、肺炎1次;4月余认生,现会发"ma ma"音;大运动发育稍落后,7个月会独坐,10个月会爬,现可扶站。出生4个月时血常规检查结果提示白细胞17.95 × 109/L(正常参考值为5.10 ~ 14.10 × 109/L),红细胞3.98 × 1012/L(正常参考值为4.00 ~ 5.50×1012/L),血红蛋白115 g/L(正常参考值为107 ~ 141 g/L),中性粒细胞占比68.2%(正常参考值为13.0% ~ 55.0%),淋巴细胞占比25.4%(正常参考值为33.0% ~ 77.0%),中性粒细胞12.23 × 109/L(正常参考值为0.80 ~ 5.80 × 109/L),淋巴细胞4.56 × 109/L(正常参考值为2.40 ~ 8.70 × 109/L),C-反应蛋白31.52 mg/L(正常参考值为0 ~ 10.00 mg/L)。尿常规检查结果提示红细胞25/高倍视野[正常参考值为(0 ~ 3)/高倍视野],白细胞146/高倍视野[正常参考值为(0 ~ 5)/高倍视野],诊断为泌尿道感染。膀胱输尿管造影检查提示双侧膀胱输尿管反流(图1),肾盂积水(分离10.8 mm,轻度)。心脏彩色多普勒超声检查提示心脏肌部室间隔缺损(图2A)、卵圆孔未闭(图2B)、左肺动脉中远端稍窄(图2C)。出生6个月头颅及腰骶部MRI检查提示白质髓鞘化落后,双侧侧脑室增宽(图3),左肾略大,肾盂周围异常信号,尾椎上翘,余腰骶椎MRI平扫未见明显异常。患儿父母表型未见异常,否认近亲结婚及家族遗传病史。患儿有1个6岁的哥哥,现体健。患儿入院时体质量为8 kg、身高为72 cm,头围44.5 cm,生长发育落后于同龄儿。精神可,有额头突出、眼窝凹陷、宽眼距、眼裂下斜、耳后旋、薄上唇、腰骶部隐窝等外观特征。肺、腹检查未见异常,四肢肌张力低下,双侧膝反射可引出。抽取患儿及其父母的外周血样,进行全外显子组测序分析,结果提示患儿染色体17p13.3区存在1.49 Mb微缺失:seq[GRCH37]del(17)(p.13.3p.13.3)chr7:g.882 518-2 371 192 del(图4),父母该区域均未发现相同的缺失。本研究通过了河南省儿童医院郑州儿童医院伦理委员会的审查(2022-K-070),患儿父母均签署了知情同意书。
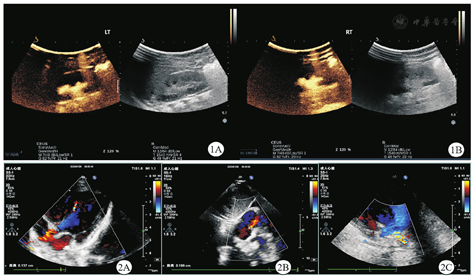
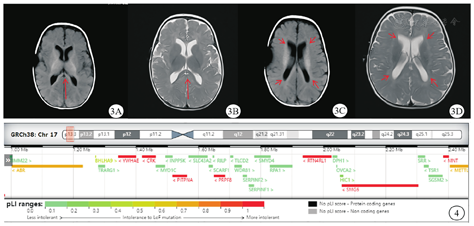
17p13.3微缺失综合征较为罕见,缺失区域包含YWHAE、CRK、HIC1等基因[1],主要临床表现包括生长发育迟缓、智力障碍、面容畸形等[2,3]。此外,Bruno等[4]曾报道17p13.3微缺失综合征患儿也会表现出膀胱输尿管反流。本例患儿具有双侧膀胱输尿管反流、生长发育迟缓、面容畸形、先天性心脏病(室间隔缺损、左肺动脉中远端稍窄,卵圆孔未闭)等临床表现,全外显子组测序发现其染色体17p13.3区存在1.49 Mb缺失片段,主要包含YWHAE、CRK、SMG6、PRPF8、HIC1等基因,故诊断为17p13.3微缺失综合征。目前国内外报道的17p13.3微缺失综合征共31例,国内仅7例,且多数在产前确诊[5]。本研究进一步丰富了国内相关病例的报道,为17p13.3微缺失综合征的诊断提供了更多的线索。
所有作者均声明不存在利益冲突

























