
版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
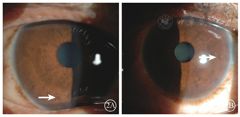
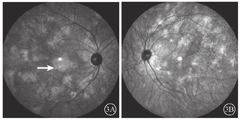
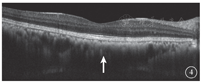
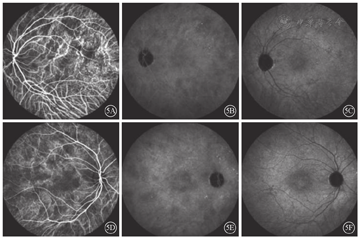
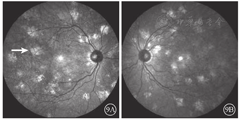
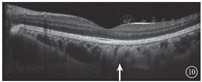
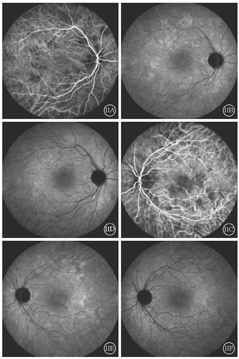
患者女,51岁,因左眼视力下降6年于2018年1月15日到吉林大学第二医院眼科就诊。否认相关既往史及家族史。患者颜面部及上肢可见多个神经纤维瘤及咖啡斑(图1)。眼科检查:右眼、左眼矫正视力分别为1.0、0.15。双眼虹膜见Lisch结节(图2),其余眼前节正常。眼底彩色照相、FAF及FFA检查,双眼均未见异常。红外眼底成像检查,双眼后极部多发片状强反射病灶(图3)。OCT增强深度成像(EDI-OCT)检查,与红外眼底成像强反射病灶对应处脉络膜毛细血管层反射增强(图4)。ICGA检查,早期可见与红外眼底成像强反射病灶相对应处呈弱荧光,随着时间延长,中晚期弱荧光面积逐渐缩小直至消失(图5)。诊断:Ⅰ型神经纤维瘤病。
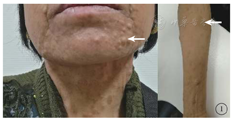
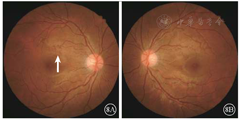
患者女,26岁,为例1患者女儿。因母亲诊断为Ⅰ型神经纤维瘤病于2018年1月16日到吉林大学第二医院眼科就诊。无异常视觉主诉,否认除母亲以外其他家人有相关症状体征。患者颜面及肢体见多个神经纤维瘤及咖啡斑(图6)。眼科检查:双眼视力1.0。双眼虹膜见Lisch结节(图7)。双眼眼前节大致正常。眼底彩色照相可见其黄斑区末梢血管略扭曲(图8)。红外眼底成像、EDI-OCT及ICGA检查表现与其母亲相似(图9,图10,图11)。诊断:Ⅰ型神经纤维瘤病。
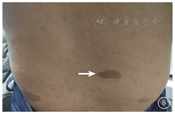
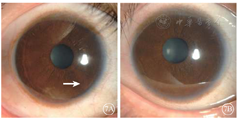
Ⅰ型神经纤维瘤病是一种常见的常染色体显性遗传病,高达50%的患者是自发突变的结果,患者可以在任何系统中发生良性或者恶性肿瘤,体表可见多发的神经纤维瘤以及牛奶咖啡斑[1]。1988年美国国立卫生研究院制定的神经纤维瘤病的诊断标准指出,在多发性皮肤咖啡斑、神经纤维瘤、腋窝或腹股沟区雀斑、视神经胶质瘤、Lisch结节、特征性骨病变、家族史中存在任何两项即可诊断为Ⅰ型神经纤维瘤病[2]。本文报道的两例患者均具有多发咖啡斑、神经纤维瘤、Lisch结节及家族史,Ⅰ型神经纤维瘤病诊断明确。Lisch结节由增生的黑色素细胞组成,紫外线可以促进其增生,随着年龄的增加,虹膜接受更多的紫外线照射,Lisch结节的数量随之增加[3]。而在Lisch结节的位置分布上,接受紫外线照射较多的下半部虹膜多于接受紫外线照射相对较少的上半部虹膜[4]。与文献报道相符,例1患者较例2患者年长25岁,可见其双眼虹膜Lisch结节数量及体积显著大于例2患者,且大多分布于下方虹膜。
Ⅰ型神经纤维瘤病大多存在脉络膜结节,常规眼底检查和眼底彩色照相不能发现病变[5]。本文例2患者仅在彩色眼底像中可见双眼黄斑区末梢血管略扭曲,这样的异常血管只存在于视网膜浅层,常见于颞上、颞下静脉的二三级分支,且在FFA上无荧光素渗漏;在EDI-OCT上可见对应位置的脉络膜毛细血管层萎缩以及脉络膜大血管缺失。但脉络膜大血管在解剖结构上是缺失还是仅仅被压缩,尚无定论。对于这样的脉络膜结节,最有效的检查手段是红外眼底成像。这些脉络膜结节由包绕神经纤维轴突增生的施万细胞和黑色素细胞组成,由于黑色素细胞对红外线的反向散射作用,病灶在红外眼底成像上表现为强反射。另外,脉络膜结节的致密结构降低了可吸收红外线的血红蛋白和水等血液成分的含量,病变区域红外线吸收得相对减少也是红外眼底成像上反射增强的原因之一[6]。
在ICGA上,由于脉络膜结节对其上方脉络膜血流的影响,早期表现为与红外眼底成像强反射区域相对应的弱荧光区。随造影时间延长,弱荧光区域的面积逐渐减小乃至消失。有文献报道,绝大多数患者脉络膜结节对应的ICGA弱荧光会随着时间延长而逐渐减小,至30 min左右逐渐消失;仅有少数年龄较大,皮下神经纤维瘤更加严重的患者,直至造影晚期,仍然存在小片弱荧光[7]。本文例1患者,年长且皮下神经纤维瘤病变严重,ICGA 16 min中期像上存在弱荧光区域,甚至在30 min晚期像上仍然遗留小片弱荧光区域;病变较轻的例2患者,16 min中期像及30 min晚期像上弱荧光区域消失。
Ⅰ型神经纤维瘤病的临床表现可从单纯的皮肤病变到在神经系统、骨骼等的完全表达,其演变以及家族间、家族内的预后难以预测,目前的治疗方法均为对症治疗。神经胶质瘤及其他典型的儿童恶性肿瘤都与Ⅰ型神经纤维瘤病有关,患有神经纤维瘤病的患者中乳腺和其他内脏肿瘤的发生率高于健康人群,部分患者可出现动静脉的狭窄及畸形。青春期为Ⅰ型神经纤维瘤病患者良性及恶性肿瘤的高发期,但神经纤维瘤、咖啡斑等相关体征却不显著[8]。患者儿童时期红外眼底成像脉络膜结节检出率远高于Lisch结节的检出率[8],对于有相关家族史或疑诊Ⅰ型神经纤维瘤病者的早期诊断有重要意义。对于Ⅰ型神经纤维瘤病,早期诊断及针对相关并发症的关注和积极随访是目前提高患者生活质量及寿命的重要手段,同时应积极进行基因诊断及产前诊断,预防后代患病。
所有作者均声明不存在利益冲突

























