
急性髓细胞白血病(AML)为异质性血液系统恶性肿瘤。AML除与相关基因突变、染色体变异相关以外,与异常的表观遗传学改变亦有着密切的关系。DNA与组蛋白甲基化修饰均为AML重要的表观遗传学改变,并且表观遗传学改变是可逆的,因此其有可能成为治疗AML的新靶点。目前,AML治疗主要采用传统的诱导化疗方案,但是疗效欠佳。为了进一步提高AML的临床疗效,亟需进一步阐明AML的发病机制,并且针对新的治疗靶点,研发新的治疗药物或创新联合药物治疗方案,以实现AML的个体化治疗。笔者拟就AML相关异常甲基化修饰的基因突变及DNA甲基化转移酶抑制剂(DNMTi)在治疗AML中的研究进展进行综述。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
急性髓细胞白血病(acute myeloid leukemia,AML)为异质性造血干细胞克隆性疾病,以细胞髓样分化严重阻滞、细胞快速克隆增殖及骨髓中未成熟的髓样细胞浸润机体的其他器官(主要为脾、淋巴结等)为主要特征。AML的发病率随年龄的增长而增高。AML被认为是成年人最常见的急性白血病,在儿童中发病率则较低,仅占儿童白血病的15%~20%[1]。某些获得性疾病可以转化为AML,最常见的是骨髓增生异常综合征(myelodysplastic syndromes,MDS)转化为AML。AML细胞分子遗传学的改变,在不同发病年龄患者中不完全相同。研究结果表明,AML的发生、发展是多因素共同作用的结果,主要与基因突变、染色体异常及表观遗传学改变相关[2]。通过对AML相关表观遗传学改变的研究,将有助于了解AML的发病机制,阐明AML潜在的治疗靶点并改善患者预后。目前,AML治疗主要采用传统的诱导化疗方案,但是疗效欠佳,为了进一步提高临床疗效,亟需进一步阐明AML的发病机制,并且针对新的治疗靶点,研发新的治疗药物或创新联合药物治疗方案,以实现AML的个体化治疗。笔者拟就AML相关异常甲基化修饰的基因突变及DNA甲基化转移酶抑制剂(DNA methyltransferase inhibitor,DNMTi)治疗AML的研究进展进行综述如下。
表观遗传学改变是指在基因核苷酸序列不发生变化的情况下,基因表达的可遗传性改变。表观遗传学改变包括DNA甲基化、组蛋白翻译后修饰及非编码RNA的调控[3]。
DNA甲基化为重要的表观遗传学改变,异常DNA甲基化可以使相关基因沉默或激活,促进AML的发生、发展[4]。DNA甲基化是在DNA甲基转移酶(DNA methyltransferase,DNMT)作用下,以S-腺苷甲硫氨酸(S-adenosyl methionine,SAM)为甲基供体,将甲基转移至胞嘧啶的过程,使得胞嘧啶第5位C被甲基化,从而转变为5-甲基胞嘧啶(5-methylcytosine,5-mC)[5]。
90%的DNA甲基化发生在CpG岛。DNA甲基化为机体正常生长、发育所必需的表观遗传学修饰,广泛参与细胞发育与分化调节等过程[6];而异常的DNA甲基化状态(甲基化水平的减低与增高)均可以导致全基因组的不稳定,从而促进肿瘤的发生、发展。病理状态下,重复序列与卫星DNA区域CpG岛呈广泛低甲基化状态,而启动子区域抑癌基因的CpG岛则呈高甲基化状态[7]。
CpG甲基化结合蛋白(methy-CpG-binding protein,MBP)在CpG岛的高甲基化,在促进AML发生、发展的作用机制中,发挥着重要的中心桥梁作用,其通过特异性识别、结合并募集辅阻遏复合物,例如组蛋白去乙酰化酶(histone deacetylase,HDAC),组蛋白甲基转移酶(histone methyltransferase,HMT)至DNA甲基化位点,引起染色质结构的修饰与重塑,使其发生浓缩、聚集,从而导致相关基因沉默[8]。目前,已发现的MBP有3类,分别为甲基化CpG结合域(methyl-CpG-binding domain,MBD)蛋白家族,Kaiso及SRA(SET and ring finger-associated)家族[1,2],其中研究最为广泛的是MBD蛋白家族,包括MBD1~6蛋白,均为甲基化依赖的转录抑制因子;此外,SET结构域分支型(SET domain bifurcated,SETDB)1,SETDB2,溴结构区邻近至锌指结构蛋白(bromodomain adjacent to zinc finger domain,BAZ)2A,BAZ2B,这4个蛋白均被认为亦具有MBD结构域[9,10]。此外,MBD蛋白亦可以通过核小体聚集及相关复合物的形成,直接参与染色质高级有序结构的组装,介导或抑制相关基因的表达[11,12]。非CpG岛的甲基化促进肿瘤发生、发展的机制尚不明确,仍需进一步研究。
基因组甲基化状态的建立及维持与DNMT密切相关。DNMT家族中具有活性的甲基转移酶为DNMT1、DNMT3a与DNMT3b,通过与HDAC结合发挥DNA甲基化修饰作用[8,13]。DNMT1可以与细胞增殖核抗原(proliferating cell nuclear antigen,PCNA)及泛素样含PHD和环指域(ubiquitin-like with PHD and ring finger domain,UHRF)1形成PCNA-UHRF1-DNMT1复合物,在特定DNA序列位点催化半甲基化DNA进行甲基化修饰,并维持细胞分裂周期中的DNA甲基化状态[14]。DNMT1基因突变可见于小部分AML患者中,并且DNMT1基因突变在T淋巴细胞、B淋巴细胞及其他骨髓恶性肿瘤患者中高表达,而在其他血液系统恶性肿瘤患者中,DNMT1基因突变极为罕见[15]。DNMT3a与DNMT3b以非甲基化的原始双链DNA为模板,催化从头甲基化(de novo methylation)[16],其基因突变多发生于第882位的精氨酸(R882),通过产生抑制DNMT3a活性的亚效等位基因蛋白,从而导致相关基因异常甲基化[17]。在正常核型急性髓细胞白血病(cytogenetically normal acute myeloid leukemia,CN-AML)患者中,DNMT3a基因突变率达30%,其可以作为AML表观遗传学治疗的靶点之一[18]。伴有DNMT3a基因突变的AML患者,通常同时伴有FLT3,核仁磷蛋白(nucleophosmin,NPM)1,异柠檬酸脱氢酶(isocitrate dehydrogenase,IDH)1/2基因突变[3],并且DNMT3a基因突变发生在上述基因突变之前,提示早期AML中DNMT3a基因突变可以导致基因染色体组结构不稳定,进而促进多基因突变,DNMT3a基因突变通常与AML不良预后相关[19]。DNMT2为RNA甲基化转移酶,DNMT2缺失对整体CpG岛甲基化水平无显著影响[20]。DNMT3L缺乏甲基转移酶活性,但是能够识别H3K4未甲基化尾部,并且通过增加DNMT3活性位点稳定性,提高DNMT3a与SAM的结合能力,从而对DNA的从头甲基化发挥重要作用[21]。
TET蛋白家族共有3个成员,分别为TET1、TET2及TET3,均为DNA羟化酶。TET家族在CpG岛将5-mC催化生成5-羟甲基胞嘧啶(5-hydroxymethylcytosine,5-hmC),5-胞嘧啶甲酰(5-formylcytosine,5-fmC)及5-胞嘧啶羧基(5-carboxyletosine,5-caC)[22],并且在碱基切除修复蛋白的作用下,移除异常DNA甲基化[23],发挥去甲基化作用。在AML患者中,TET2基因突变率为7%~23%,并且在CN-AML患者中其基因突变率较其他AML患者更高,通常与老龄、高白细胞计数、低血小板计数、NPM1与ASXL1基因突变相关[24]。TET2基因突变对AML患者临床结局的影响尚存在争议[25,26],这可能与基因突变位点不同,或有其他基因突变共同作用相关。
IDH家族为烟酰胺腺嘌呤二核苷酸磷酸(nicotinamide adenine dinucleotide phosphate,NADP)依赖性酶,在三羧酸循环中以烟酰胺腺嘌呤二核苷酸(nicotinamide adenine dinucleotide,NAD)或NADP作为辅因子,催化异柠檬酸氧化脱酸生成α-酮戊二酸(α-ketoglutaric acid,α-KG),同时生成还原型烟酰胺腺嘌呤二核苷酸(reduced nicotinamide adenine dinucleotide,NADH)或者还原型烟酰胺腺嘌呤二核苷酸磷酸(reduced nicotinamide adenine dinucleotide phosphate,NADPH),参与细胞内氧化损伤防御机制[27]。突变型IDH基因失去正常的催化能力,使得NADPH与α-KG结合,催化α-KG转化为D型2-羟基戊二酸(2-hydroxyglutaric acid,2-HG),即D-2-HG,从而消耗α-KG,竞争性抑制α-KG依赖性酶,例如TET2酶与组蛋白赖氨酸去甲基化酶(K-demethylase,KDM),导致DNA、组蛋白异常甲基化,进而促进AML的发生、发展[28]。IDH1/2基因突变通常表现为杂合突变,例如IDH1基因突变位点为R132,IDH2基因突变位点为R140与R172[27]。目前研究认为,伴有不同IDH基因突变的AML患者预后不同,例如伴有IDH2 R140基因突变的AML患者预后良好,伴有IDH2 R172基因突变者则相反[29]。CN-AML患者的IDH基因突变率高于其他AML患者,IDH1与IDH2基因突变率分别为10%~16%、10%~19%。此外,IDH1与IDH2基因突变同时出现的情况极少[30]。伴有IDH基因突变的AML患者中,2-HG高表达,这提示血清IDH水平可以作为疾病活动监测指标,并且作为治疗反应的生物学标志物,用于预测疾病预后。
组蛋白修饰包括乙酰化、甲基化、泛素化、磷酸化修饰。甲基化修饰为组蛋白修饰的重要方式,其中甲基化位点多位于组蛋白H3、H4的赖氨酸与精氨酸残基上[31]。组蛋白甲基化对基因转录的影响取决于甲基化残基位置与甲基化程度,例如H3K4、H3K36、H3K79的甲基化与基因转录激活相关,而H3K9、H3K27的甲基化与基因转录抑制相关。研究发现,组蛋白甲基化修饰对DNA甲基化起促进与协同作用,这提示DNMTi与KDM联合使用,可能是合理治疗AML的策略之一[32]。
MLL基因位于染色体11q23,MLL蛋白具有HMT活性,介导基因转录调控,激活相关的表观遗传学修饰,并且参与HOX基因的转录调控。AML患者中MLL基因发生部分串联重复(partial tandem duplication,PTD)的突变率达5%~7%,这与AML患者的不良结局相关[33]。另外,MLL基因位点常发生染色体易位与重排,可以产生诸多融合蛋白,例如HMT-DOT1L(disruptor of telomeric silencing 1-like)蛋白,从而促进H3K79甲基化[34]。MLL基因发生PTD突变及MLL基因所在染色体异位与重排,均能够通过使HOX基因表达抑制,引起造血干细胞发育异常,促进AML的发生、发展。
ASXL基因为PcG(polycomb group)与TrxG(trithorax group)基因的增强子,有3个同源基因,分别为ASXL1、ASXL2、ASXL3,其中AML患者的ASXL1基因突变率为6%~30%,伴有ASXL1突变者预后较差,并且多有MDS病史[35]。ASXL1基因通过与多梳蛋白抑制复合物(polycomb repressor complex,PRC)2共同作用,参与DNA表观遗传学调控,对具有基因转录抑制作用的H3K27进行三甲基化作用生成H3K27me2,因此ASXL1基因突变将导致H3K27甲基化水平减低,从而减低H3K27对于相关基因的转录抑制作用[36]。
DNA甲基化为可逆的过程。利用去甲基化药物可以通过抑制启动子区域CpG岛甲基化,激活沉默的抑癌基因,达到治疗AML的目的,为目前治疗AML的研究思路。临床上,常用的去甲基化药物为DNMTi。根据化学结构,DNMTi可分为核苷类与非核苷类DNMTi,其中非核苷类DNMTi不整合入DNA及RNA,细胞毒性较小,例如普鲁卡因、As2O3等[40]。核苷类DNMTi则包括阿扎胞苷、地西他滨等。
目前,阿扎胞苷及其脱氧类似物地西他滨为DNMTi中研究最为透彻的2种胞苷类似物,并在临床上广泛应用于血液系统恶性疾病,特别是用于MDS的预防与治疗。阿扎胞苷在体内三磷酸化生成三磷酸阿扎胞苷,其中80%~90%三磷酸阿扎胞苷整合入RNA结构中,影响细胞核与细胞质中的RNA相关代谢活动,包括影响核糖体生物合成与蛋白质的合成等[40,41,42]。此外,10%~20%三磷酸阿扎胞苷转化为脱氧三磷酸阿扎胞苷,在DNA复制过程中整合入DNA结构中,取代第6位胞嘧啶与DNMT形成共价键[41,43]。地西他滨作用机制与阿扎胞苷相似,但是仅整合入DNA结构中。
阿扎胞苷与地西他滨于20世纪60年代被首次合成,但是大剂量药物治疗的强烈细胞毒性作用及骨髓抑制作用,限制了阿扎胞苷与地西他滨在临床上的应用[44]。随后地西他滨被证实剂量反应曲线呈U形,即具有剂量效应,低剂量时主要通过抑制DNMT作用影响DNA甲基化,而高剂量时通过渗入细胞中发挥细胞毒性作用[45]。这表明在临床上使用低剂量阿扎胞苷与地西他滨,可以达到有效性与安全性的统一。研究认为,阿扎胞苷与地西他滨在临床应用中具有良好应答,主要通过减低DNA甲基化水平,从而激活沉默的抑癌基因及使肿瘤抗原表达水平增高,以增加肿瘤免疫反应发挥作用[46,47]。然而目前的临床证据不完全支持阿扎胞苷及地西他滨治疗AML的临床应答与阿扎胞苷及地西他滨的低甲基化诱导作用之间存在直接相关性。另外部分研究发现,地西他滨在细胞核中通过泛素依赖性激活蛋白酶体途径选择性破坏DNMT1功能,从而达到治疗目的[48]。阿扎胞苷与地西他滨的药物作用机制,尚需要进一步的研究予以证实,以最大限度地提高其临床效用。
其他核苷类DNMTi包括:①guadecitabine(SGI-110),地西他滨与脱氧鸟苷合成的新型低甲基化核苷酸,具有耐胞苷脱氨酶的活性,可以增加地西他滨在体内稳定性及延长药物半衰期,提高地西他滨的生物利用度,目前处于治疗AML与MDS的Ⅱ期临床试验阶段[49]。②zebularine,阿扎胞苷的衍生物,于1961年被首次合成,并被证实具有胞苷脱氨酶活性与抗肿瘤作用,随后研究发现其具有DNA去甲基化作用,并且能够重新激活肿瘤抑制基因。zebularine具有良好的药代动力学特征,细胞毒性较阿扎胞苷及地西他滨低,因此可以长期给药,以防止甲基化再激活。zebularine的作用机制迄今尚不明确,需要进一步研究,目前处于临床前期试验阶段[50]。
癌症和白血病B协作组(Cancer and Leukemia Group B,CALGB)进行的编号为CALGB 8421的Ⅱ期临床试验结果显示,研究纳入的43例接受阿扎胞苷持续静脉滴注的AML与MDS患者中,完全缓解(complete remission,CR)率为12%(5/43),部分缓解(partial remission,PR)率为25%(11/43),中位生存期为14.7个月[51]。随后编号为CALGB 8912的临床试验纳入68例AML与MDS患者接受阿扎胞苷皮下注射,研究结果显示MDS患者CR率为12%(8/68),PR率为15%(10/68)[51]。编号为CALGB 9221的临床试验,采用静脉滴注或者皮下注射阿扎胞苷对99例AML与MDS患者进行治疗,研究结果显示,患者总体缓解率(overall response rate,ORR)为23%(23/99),中位生存期为21个月[51]。该研究认为,阿扎胞苷能够提高MDS与AML患者的生存质量,延长患者的总体生存(overall survival,OS)期。在上述临床试验结果的支持下,2004年美国食品药品监督管理局(Food and Drug Administration,FDA)批准阿扎胞苷作为治疗MDS的一线治疗药物[52]。AZA-001试验中,比较骨髓原始细胞比例达到20%~30%的MDS患者(根据最新世界卫生组织分类诊断标准诊断为AML患者)接受阿扎胞苷与传统治疗方案的疗效,与接受传统治疗方案的患者相比,接受阿扎胞苷治疗者能够获得更长的中位生存期(24.5个月比15.0个月,P=0.01),更高的2年OS率(50.0%比26.0%,P<0.01),血液学缓解持续时间获得延长(13.6个月比5.2个月,P<0.01),并且能够一定程度减低依赖性输血的发生率(11.4%比45.0%,P<0.05)[53]。此外,2008年美国癌症综合网络(National Comprehensive Cancer Network,NCCN)AML治疗指南针首次将阿扎胞苷低强度治疗方案,纳入无严重并发症、年龄>60岁的AML患者的治疗选择[54]。
编号为D-0007的临床随机对照试验,将179例MDS患者随机分为采用西他滨15 mg/m2,连续静脉输注8 h,应用3 d,每6周1次的治疗组(n=89)与采用最佳支持治疗的对照组(n=80),该研究结果显示,治疗组ORR为30%(27/89),其中CR率为9%(8/89),PR率为8%(7/89),而对照组ORR为7%(6/80),2组ORR相比,差异有统计学意义(P<0.001);治疗组中位持续反应期为10.3个月[55]。进一步研究结果显示,对于初发患者,治疗组MDS患者转化为AML或者死亡的中位时间较对照组延长(12.6个月比9.4个月,P<0.05),治疗组高危MDS患者转化为AML或死亡的中位时间较对照组亦延长(9.3个月比2.8个月,P<0.05)[55]。在上述临床试验结果支持下,美国FDA于2006年批准地西他滨用于MDS患者的治疗,并且2012年欧洲委员会(European Union,EU)亦批准地西他滨单药用于治疗初治AML老年患者。
为确定DNMTi诱导DNA去甲基化的最低有效剂量并达到最佳临床效果,1项Ⅰ期临床试验纳入48例血液系统恶性疾病患者,其中44例为AML与MDS患者,3例为慢性髓细胞白血病(chronic mylogenous leukemia,CML),1例为急性淋巴细胞白血病(acute lymphocytic leukemia,ALL),全部患者静脉滴注地西他滨5~20 mg/(m2·d),连续应用10~20 d后,评估不同剂量地西他滨的疗效差异[56]。该研究结果显示,接受静脉滴注地西他滨15 mg/(m2·d),连续应用10 d治疗方案患者的临床应答率最高(65%,11/17)[56]。基于上述临床试验结果,为了对地西他滨治疗剂量与方案进行进一步探讨,多项Ⅱ/Ⅲ期临床试验陆续进行。其中,Kantarjian等[57]纳入95例AML与MDS患者,并且随机给予3种地西他滨治疗方案,观察患者疗效。该研究中,患者接受的3种治疗方案包括:①静脉滴注地西他滨20 mg/(m2·d),连续应用5 d;②皮下注射地西他滨20 mg/(m2·d),连续应用5 d;③静脉滴注地西他滨10 mg/(m2·d),连续应用10 d;上述治疗方案每4周进行1次。该研究中位治疗疗程数为6次,治疗后患者的总体反应率为73%(69/95),CR率为34%(32/95),其中接受5 d静脉滴注方案治疗者的CR率最高,为39%(25/64),5 d皮下注射者的CR率为21%(3/14),10 d静脉滴注者的CR率为24%(4/17),三者相比,差异有统计学意义(P<0.05)[57]。该研究结果提示,在治疗MDS或AML患者时,减低地西他滨总剂量与延长给药时间可能使患者受益[57]。部分DNMTi治疗AML与MDS的临床试验结果[51,53,55,57],见表1。

DNMTi治疗AML与MDS的部分临床试验结果
DNMTi治疗AML与MDS的部分临床试验结果
| 研究者 | 例数 | 疾病类型 | 治疗方案 | 疗效 |
|---|---|---|---|---|
| Silverman等[51] | 99 | AML与MDS | 静脉滴注或皮下注射阿扎胞苷75 mg/(m2·d),连续应用7 d,28 d为1个周期 | ORR为23%,中位生存期为21个月 |
| Fenaux等[53] | 358 | AML与MDS | 静脉滴注或皮下注射阿扎胞苷75 mg/(m2·d),连续应用7 d,28 d为1个周期 | OS率为50%,中位生存期为24.5个月 |
| Kantarjian等[55] | 170 | MDSa | 静脉滴注地西他滨15 mg/(m2·d),连续应用3 d,每6周应用1次 | ORR为30%,其中CR率为9%,PR率为8% |
| Kantarjian等[57] | 95 | AML与MDS | 静脉滴注或皮下注射地西他滨10 mg/(m2·d)或者20 mg/(m2·d),连续应用5 d或者10 d | ORR为73%,CR率为34% |
注:a根据AML最新世界卫生组织分类诊断标准,应该诊断为AML。DNMTi为DNA甲基化转移酶抑制剂,AML为急性髓细胞白血病,MDS为骨髓增生异常综合征,ORR为总体缓解率,OS为总体生存,CR为完全缓解,PR为部分缓解
随着以DNMTi为代表的靶向表观遗传学药物的问世,该类药物通过DNA去甲基化作用,应用于MDS与AML患者的临床治疗,并且取得了一定疗效,使AML与MDS患者预后得到改善。DNMTi联合其他具有促进或协同作用的药物,进一步提高AML患者临床疗效,已成为治疗AML的新研究方向。DNMTi的协同药物之一是组蛋白去乙酰化酶抑制剂(histone deacetylase inhibitor,HDACi),包括丙戊酸、帕比司他、伏林司他等[58]。抑癌基因启动子甲基化与组蛋白去乙酰化的表观遗传学改变所导致的基因沉默,为AML发生、发展的分子机制之一,这为DNMTi与HDACi联合使用提供理论依据。
DNMTi与传统化疗药物联合应用亦是AML治疗的研究热点之一。在治疗AML中,DNMTi与细胞毒性药物之间具有协同作用的可能原因包括:①DNMTi通过DNA去甲基化作用,重新激活沉默的抑癌基因和(或)化疗敏感的基因,使肿瘤细胞对细胞毒性化疗药物更为敏感。②DNMTi治疗可以导致广泛的DNA与组蛋白低甲基化,因而导致表观遗传学修饰位点的改变,同时广泛的DNA低甲基化状态与细胞染色体不稳定性相关,可以增强细胞毒性药物的DNA损伤作用,达到治疗目的[58,59,60,61]。
近年,国内外已经开始进行DNMTi联合传统化疗药物在AML患者中的小样本临床试验。地西他滨联合传统化疗药物治疗老年、不适合大剂量化疗及复发/难治性AML患者的疗效显著。部分地西他滨联合传统化疗药物治疗AML与MDS的临床试验结果[58,59,60,61],见表2。
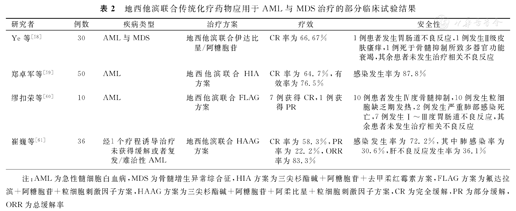
地西他滨联合传统化疗药物应用于AML与MDS治疗的部分临床试验结果
地西他滨联合传统化疗药物应用于AML与MDS治疗的部分临床试验结果
| 研究者 | 例数 | 疾病类型 | 治疗方案 | 疗效 | 安全性 |
|---|---|---|---|---|---|
| Ye等[58] | 30 | AML与MDS | 地西他滨联合伊达比星/阿糖胞苷 | CR率为66.67% | 1例患者发生胃肠道不良反应,1例发生Ⅱ级皮肤瘙痒,1例死于骨髓抑制所致多器官功能衰竭,其余患者未发生治疗相关不良反应 |
| 郑卓军等[59] | 50 | AML | 地西他滨联合HIA方案 | CR率为64.7%,有效率为76.5% | 感染发生率为87.8% |
| 缪扣荣等[60] | 10 | AML | 地西他滨联合FLAG方案 | 7例获得CR,1例获得PR | 10例患者发生Ⅳ度骨髓抑制,10例发生粒细胞缺乏期发热,2例发生严重肺部感染死亡,7例发生Ⅰ~Ⅲ度胃肠道不良反应,其余患者未发生治疗相关不良反应 |
| 崔巍等[61] | 36 | 经1个疗程诱导治疗未获得缓解或者复发/难治性AML | 地西他滨联合HAAG方案 | CR率为58.3%,PR率为22.2%,ORR率为83.3% | 感染发生率为72.2%,其中肺感染率为30.6%,肝不良反应发生率为36.1% |
注:AML为急性髓细胞白血病,MDS为骨髓增生异常综合征,HIA方案为三尖杉酯碱+阿糖胞苷+去甲柔红霉素方案,FLAG方案为氟达拉滨+阿糖胞苷+粒细胞刺激因子方案,HAAG方案为三尖杉酯碱+阿糖胞苷+阿柔比星+粒细胞刺激因子方案,CR为完全缓解,PR为部分缓解,ORR为总缓解率
DNMTi与传统细胞毒性化疗药物联合治疗AML患者具有良好前景,但是目前相关临床试验多为Ⅰ/Ⅱ期临床试验,样本量较少,临床试验异质性较大,以成年人复发/难治性AML与MDS的临床研究为主,同时这2类药物联合应用的作用机制尚不清楚,临床应用方案等仍需要进一步研究,以明确这2类药物联合应用的确切作用机制、疗效、联合用药方案与最佳治疗剂量等问题,以更好的通过联合不同药物达到最佳疗效。
表观遗传学改变及与甲基化相关的基因突变在AML的发生、发展中的作用,越来越引起相关研究者的重视。全基因组甲基化测序与分析方法的发展,使研究者对全基因组DNA与组蛋白甲基化有了更深入了解。然而,目前仍有诸多问题亟需解决,例如表观遗传学改变与遗传改变如何共同作用导致AML的发生、发展,甲基化改变能否作为AML预后判断、诊断分型、疗效评估、疾病控制等的新观察指标等。此外,笔者阐述了以DNMTi为代表的表观遗传学药物的临床应用进展,但是靶向表观遗传学药物的体内作用机制、最佳治疗剂量、联合用药方案等问题仍需进一步研究。表观遗传学改变是可逆的,因此系统、全面、深入地研究AML的发病机制,有可能为AML患者临床诊治、预后判断提供新的依据,并可以成为新的治疗靶点,真正实现AML患者的个体化治疗。
无

























