
静脉血栓栓塞症(VTE)是由遗传和环境因素共同引起的疾病。与欧洲和北美洲人群常见的遗传危险因素不同,亚洲人群VTE的主要遗传危险因素是遗传性抗凝蛋白缺乏。蛋白C和蛋白S均为维生素K依赖的抗凝蛋白,在调节机体凝血功能过程中发挥重要作用。因此,遗传性蛋白C和蛋白S缺乏症相关遗传危险因素的鉴定,对亚洲人群VTE的临床研究具有重要意义。近年来,相关研究已明确部分亚洲人群中遗传性蛋白C和蛋白S缺乏症常见的PROC和PROS1基因突变,并证实其与VTE发生风险增加密切相关。为了提高对VTE的诊治水平,笔者拟就亚洲人群中遗传性蛋白C和蛋白S缺乏症的常见遗传危险因素进行阐述。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
近年来,由于静脉血栓栓塞症(venous thromboembolism,VTE)的发病率及其导致的患者病死率逐年上升,该病越来越受到临床重视。VTE是一种受多种因素共同影响的复杂疾病,其中遗传危险因素在其发病过程中发挥关键作用[1,2]。导致VTE的遗传危险因素多样,其中遗传性凝血因子缺陷,如凝血因子FⅤ Leiden突变(R506Q)和凝血酶原突变(G20210A),是欧洲和北美洲人群血栓形成的主要易感因素,但在亚洲人群中少见。而遗传性蛋白C、蛋白S及抗凝血酶(antithrombin,AT)等抗凝蛋白的缺乏在欧洲和北美洲人群中少见,却是亚洲人群VTE的主要遗传危险因素[3,4]。
蛋白C和蛋白S都是活化蛋白(activated protein,AP)C抗凝系统的重要组成成分。APC抗凝系统作为人体生理性抗凝系统之一,其调节失衡易造成血栓形成。APC抗凝系统功能障碍是导致亚洲人群VTE的主要遗传危险因素之一[5]。在亚洲人群中鉴定遗传性蛋白C和蛋白S缺乏症的遗传危险因素,对VTE相关临床研究具有重要意义。近年来,对遗传性蛋白C和蛋白S缺乏症的相关基础研究已取得较大进展,目前已经明确多种与APC抗凝系统功能障碍有关的常见基因突变和基因多态性。为了进一步提高VTE的治疗水平,笔者拟就亚洲人群中遗传性蛋白C和蛋白S缺乏症的常见遗传危险因素进行阐述如下。
蛋白C主要在肝内合成,是一种依赖于维生素K的丝氨酸蛋白酶原。蛋白C由染色体2q14.3上的PROC基因编码,该基因全长约为11.1 kb,包含9个外显子[6]。成熟的蛋白C由41 kD的重链和21 kD的轻链通过二硫键连接,并在外周血中循环。其多肽结构域包括轻链N端γ-羧基谷氨酸残基(γ-carboxyglutamic acid,Gla)结构域,2个表皮生长因子(epidermal growth factor,EGF)样结构域,连接肽和重链C端丝氨酸蛋白酶的催化结构域。蛋白C通过凝血酶-血栓调节蛋白复合物(thrombin-thrombomodulin compound,T-TM)被激活为APC。APC在辅助因子蛋白S、FⅤ的作用下,通过蛋白水解作用使活化的FⅤa和FⅧa失活,从而发挥抗凝作用。而内皮细胞蛋白C受体(endothelial protein C receptor,EPCR)在内皮表面与蛋白C结合后,则加速T-TM介导的APC活化[7]。此外,APC通过与EPCR结合,激活蛋白酶激活受体(protease-activated receptor,PAR)1实现细胞保护活性,包括抗凋亡活性、抗炎活性、基因表达谱的改变,以及对内皮屏障功能的稳定作用[8]。
蛋白S是APC抗凝系统中另一种重要的生理性抗凝蛋白。其主要在肝内合成,是一种维生素K依赖性非酶性血浆糖蛋白。成熟的蛋白S是由635个氨基酸组成的单链蛋白,其分子结构包含N端的Gla结构域,特异性凝血酶敏感区域(thrombin-sensitive region,TSR),4个EGF样结构域,以及1个性激素结合球蛋白(sex hormone-binding globulin,SHBG)样结构域。在血浆中约60%的蛋白S与C4b结合蛋白(C4b binding protein,C4BP)非共价结合,40%为具有辅助因子活性的游离蛋白S。蛋白S作为APC的非酶辅助因子可加速APC对FⅤa和FⅧa的蛋白水解作用,也可以作为组织因子途径抑制剂(tissue factor pathway inhibitor,TFPI)的辅助因子加速抑制FⅩa,从而在调节生理抗凝功能中发挥重要的作用[9]。此外,蛋白S通过与凋亡细胞表面表达的磷脂酰丝氨酸结合,促进早期凋亡细胞的清除[10]。
蛋白S由染色体3q11.2着丝粒附近的PROS1基因编码。PROS1基因长度约为80 kb,由15个外显子和14个内含子组成。其与同样在3号染色体着丝粒附近的转录沉默假基因PROS2的序列相似性约为97%,但是PROS2基因包含了多个编码错误,如缺失外显子1和存在部分碱基改变[11]。此外,蛋白S具有游离和结合2种形式,具有抗凝作用,却无酶促活性,这些因素均使得遗传性蛋白S缺乏症的诊断更为困难和复杂[11]。总之,蛋白C和蛋白S在调节抗凝功能中发挥重要作用。
遗传性蛋白C缺乏症是PROC基因突变引起的常染色体遗传性疾病。大多数蛋白C缺乏症患者由PROC基因杂合突变引起,患者通常无明显症状,但VTE发生风险增加。PROC双等位基因突变(纯合突变或复合杂合突变)引起的遗传性蛋白C缺乏症是罕见且危及生命的疾病,患者伴有严重血栓形成的表现,包括新生儿暴发性紫癜和弥散性血管内凝血(disseminated intravascular coagulation,DIC)[12]。然而,也有研究发现,蛋白C水平极低或者低于检测范围下限的新生儿,其临床症状发作时间较晚[6]。根据遗传性蛋白C缺乏症患者的蛋白C活性和抗原水平,可将其可分为2种类型:Ⅰ型为蛋白C活性和抗原水平均降低,该类型较常见;Ⅱ型为蛋白C抗原水平正常而活性降低。其中,Ⅱ型遗传性蛋白C缺乏症又分为2类:Ⅱa型为蛋白C酰基水解和抗凝活性均降低;Ⅱb型则为抗凝活性降低,但是酰胺水解活性正常。Ⅰ型遗传性蛋白C缺乏症最为常见,而Ⅱa型蛋白C缺乏症患者所占比例约为24.00%,Ⅱb型则十分罕见[13]。目前,在人类基因突变数据库(human gene mutation database,HGMD)中已记录416种与遗传性蛋白C缺乏症相关的PROC基因突变(http://www.hgmd.cf.ac.uk/ac)。这些突变大多数是错义突变、无义突变或者剪切突变。
遗传性蛋白S缺乏症是由PROS1基因突变引起的常染色体显性遗传病,也是亚洲人群中VTE患者的主要遗传危险因素之一。遗传性蛋白S缺乏症患者的PROS1基因杂合突变与其VTE发生风险增加相关,伴PROS1基因纯合突变的遗传性蛋白S缺乏症则可导致新生儿暴发性紫癜。在亚洲人群中遗传性蛋白S缺乏症发病率为0.06%~2.02%,VTE患者为8.00%~47.06%[5]。遗传性蛋白S缺乏症可分为3型:Ⅰ、Ⅱ和Ⅲ型遗传性蛋白S缺乏症。Ⅰ和Ⅲ型遗传性蛋白S缺乏症患者的主要特征为蛋白S总抗原和游离蛋白S抗原水平降低(Ⅰ型)和仅游离蛋白S抗原水平降低(Ⅲ型);Ⅱ型遗传性蛋白S缺乏症患者表现为蛋白S功能缺陷,其特征在于蛋白S总抗原和游离蛋白S抗原水平正常,但是蛋白S活性降低。约95%患者为Ⅰ和Ⅲ型遗传性蛋白S缺乏症,突变位点在整个PROS1基因均可见,而蛋白S缺陷(Ⅱ型缺陷)仅占5%,突变位点通常位于EGF样结构域[14]。蛋白S缺乏症具有遗传异质性,目前在HGMD中已登记了444种不同的PROS1基因突变,其中绝大多数是错义突变、无义突变和小片段缺失,仅少数是大片段缺失或者重复突变。遗传性蛋白C和蛋白S缺乏症均易引起VTE,但是具体的遗传危险因素仍需进一步明确。
目前,亚洲人群中VTE的遗传学背景尚不明确。相关研究对发生血栓患者的遗传性蛋白C和蛋白S缺乏症的患病率进行了调查。文献报道,85例日本VTE患者的PROC基因突变频率(9.00%)约为欧洲和北美洲患者(2.00%~4.00%)的3倍,PROS1基因突变频率(22.00%)比欧洲和北美洲患者(2.00%~4.00%)高5~10倍[15]。针对韩国人群抗凝蛋白缺乏症发生率和基因突变谱的研究结果显示,71例经基因测序证实为遗传性抗凝蛋白缺乏症的患者中,遗传性蛋白C缺乏症最常见(51%)[3]。而在韩国另一项研究则发现,VTE患者中遗传性蛋白质S缺乏症最常见(56%,31/55)[16]。然而,上述研究在患者中均未检测到FⅤ Leiden突变。上述研究结果表明,与欧洲和北美洲人群遗传易感因素不同,亚洲人群VTE的重要危险因素是遗传性蛋白C和蛋白S抗凝功能异常。
此外,部分调查研究结果证实,多种与抗凝蛋白活性降低和VTE风险增加有关的基因突变及基因多态性,如在日本人群中特异性的PROS1 p.Lys196Glu和中国人群中常见的PROC p.Arg189Trp基因突变。进一步了解这些遗传因素对抗凝蛋白结构和功能的影响,有助于VTE的临床诊断和预防。这些常见基因突变可为遗传性蛋白C和蛋白S缺乏症基因治疗提供新的靶点。
在对中国人群遗传性蛋白C缺乏症患者遗传学背景的调查中,Tang等[17]发现,PROC c.565C>T(p.Arg189Trp)基因突变不仅是遗传性蛋白C缺乏症的重要遗传危险因素之一,也是中国人群发生VTE的重要遗传危险因素之一。既往对中国VTE患者的PROC基因突变研究中也得出同样的结果[18]。该调查研究结果显示,该基因突变约占中国VTE患者的0.87%(9/1 031),并且与VTE发生密切相关。伴PROC p.Arg189Trp基因杂合突变的患者表现为蛋白C活性降低和抗原水平正常,主要与Ⅱ型遗传性蛋白C缺乏症有关,这与既往研究结果一致[17]。为了进一步了解该基因突变的致病机制,Ding等[19]采用哺乳动物细胞表达PROC p.Arg189Trp基因突变,以分析蛋白C的抗凝和抗炎功能的结果显示,携带该基因突变的蛋白C在没有辅助因子的情况下,具有正常的酰胺水解和蛋白水解活性,但是其与EPCR的结合亲和力降低2/3。此外,该错义突变在轻链C端的EGF-2结构域和丝氨酸蛋白酶结构域间,该区域功能尚未明确,可能影响蛋白C与其他生物分子(T-TM、蛋白S和磷脂等)的相互作用,具体影响需要进一步研究阐明。
Tang等[20]针对中国人群的VTE遗传因素分析发现,PROC c.574_576del(p.Lys193del)基因多态性是中国人群血栓形成的另一个重要遗传危险因素。该研究发现,中国人群PROC p.Lys193del基因多态性发生率为2.42% (25/1 031),低于FⅤ Leiden(5.00%~10.00%),与凝血酶原G20210A基因突变相似(2.00%~4.00%),并且高于PROC p.Arg189Trp基因突变(0.87%)[21]。但是,相关研究中,PROC p.Lys193del基因突变率的报道不一。既往研究报道,对中国23例遗传性蛋白C缺乏症患者进行遗传分析发现,PROC p.Lys193del基因多态性发生率为13.90%(5/36),但这可能是纳入患者和检查方法存在差异所致结果[18]。相关研究在体外细胞中表达该基因多态性进一步确定其对蛋白C功能的影响。Tang等[20]研究发现,突变型蛋白C的抗凝活性是野生型蛋白C的43.60%,但是酰胺分解活性相对正常。另一项研究则发现,在没有辅助因子的情况下,突变型蛋白C具有正常的酰胺水解和蛋白水解活性,在存在蛋白S的情况下抗凝活性下降30%~50%[19]。研究结果显示,PROC p.Lys193del基因多态性携带者的蛋白C抗凝活性也明显降低,与上述研究结果一致[20]。总之,PROC p.Lys193del基因多态性被认为是导致Ⅱ型遗传性蛋白C缺乏症的遗传危险因素之一,并且与蛋白C抗凝活性降低和VTE风险增加有关。
针对韩国抗凝蛋白缺乏症患者的研究结果显示,遗传性蛋白C缺乏症最常见,约占所筛查VTE患者人数的50.00%(36/71)。其中,p.Arg211Trp和p.Met406Ile错义突变占所有PROC基因突变的53.50%[分别为32.60%(14/43)和20.90%(和9/43)][3]。在抗凝蛋白缺乏症遗传危险因素筛查中,PROC Arg211Trp基因突变更占优势,而Met406Ile基因突变未检测到。这2种基因突变最初在日本VTE患者被发现可导致Ⅰ型遗传性蛋白C缺乏症[22]。在分子结构上,PROC p.Arg211Trp是在重链α-凝血酶裂解位点的CpG热点处发生突变,可能影响蛋白C折叠功能和稳定性。而PROC p.Met406Ile是在丝氨酸蛋白酶结构域的非CpG位点发生突变,这可能影响蛋白C的正常合成或者稳定性。目前,PROC p.Arg211Trp和p.Met406Ile基因突变对蛋白C功能影响的研究较少,仍需进一步探索其具体机制。
在日本VTE成年患者中,遗传性蛋白S缺乏症是最常见的遗传易感因素之一。PROS1 c.586A>G(p.Lys196Glu)基因突变在日本遗传性蛋白S缺乏症患者中很普遍,5.88%~10.00%的VTE患者伴该基因突变[15,23]。日本的多项病例对照研究结果均证实,该基因突变是VTE的遗传危险因素(OR= 3.74~8.56)[15,23]。携带PROS1 p.Lys196Glu基因突变小鼠的血浆APC辅助因子活性降低,比野生型小鼠更容易形成静脉血栓[24]。该错义突变位于蛋白S的第2个EGF样结构域,其功能障碍的机制除上述的APC辅助因子活性降低外,既往研究还发现异常蛋白S不能与FⅩa相互作用,使得其抑制凝血酶原酶复合物的能力降低[25]。但是体外哺乳动物细胞的研究结果表明,伴PROS1 p.Lys196Glu基因突变不影响TFPI辅助因子的活性,也不影响蛋白S与C4BP的结合或者凝血酶依赖性裂解引起的蛋白S功能丧失,其可能仅由于APC辅助因子活性降低而增加了血栓形成风险[26]。PROS1 p.Lys196Glu基因突变是日本VTE患者常见的遗传危险因素,但在其他国家暂未被报道。
引起亚洲人群发生VTE的抗凝蛋白基因突变频率和类型具有差异性。在日本约50.00%先天性VTE患儿表现为遗传性蛋白C缺乏症,其中大部分患者携带PROC基因纯合或者复合杂合突变,这在以PROC基因杂合突变为主的VTE日本成年患者中很少见[27,28]。对中国华南地区人群的PROS1基因突变谱相关研究则发现,PROS1 c.-168C>T和c.1680T>A基因突变较为常见[29]。Li等[30]在中国53例遗传性蛋白S缺乏症患者中首次鉴定出3个不同于其他地区人群的PROS1热点突变,即p.Glu67Ala、p.Arg561Trp及p.Tyr560Ter。这可能与"奠基者效应"引起的特定人群普遍性突变有关。这些遗传因素引起其他亚洲人群血栓形成的风险需进一步研究和评估,以提高VTE的诊断和预防水平。例如,中国人群常见的PROC p.Arg189Trp基因突变也是其他亚洲人群VTE的常见遗传危险因素[31]。但是,目前尚无在中国和韩国人群发现日本人群中常见的PROS1 p.Lys196Glu基因突变的相关报道。此外,为了更好地了解遗传因素对VTE的影响,有待对其他遗传因素进行进一步探索,以期为降低血栓形成的风险提供理论依据。例如,血栓调节蛋白(thrombomodulin,TM)基因THBD c.-151G>T多态性被发现与TM的5′非编码区功能受损和VTE风险增加有关,该杂合突变携带者发生VTE的风险为不伴该基因杂合突变者的2.80倍(95%CI:1.48~5.32倍)[32]。全基因组关联研究(genome wide association study,GWAS)和转录组范围关联研究(transcriptome-wide association studies ,TWAS)也已鉴定出新的VTE相关基因座,如C1orf198、PLEK、OSMR-AS1、NUGGC/SCARA5[33]。亚洲人群中VTE患者的遗传性蛋白C和蛋白S缺乏症相关遗传危险因素总结[3,15,17,20,23,32],见表1。总之,亚洲人群VTE的遗传因素存在差异性,需进一步鉴定患者的具体遗传变异,以全面了解VTE的遗传特征。
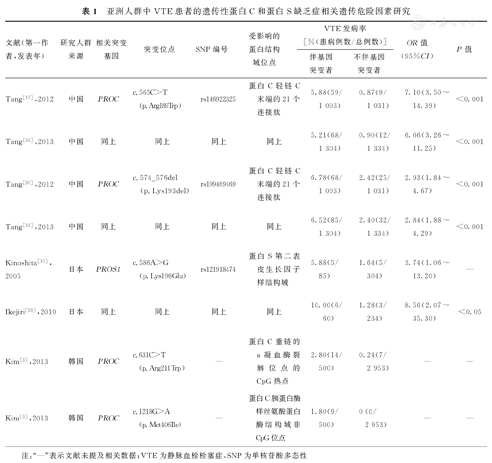
亚洲人群中VTE患者的遗传性蛋白C和蛋白S缺乏症相关遗传危险因素研究
亚洲人群中VTE患者的遗传性蛋白C和蛋白S缺乏症相关遗传危险因素研究
| 文献(第一作者,发表年) | 研究人群来源 | 相关突变基因 | 突变位点 | SNP编号 | 受影响的蛋白结构域位点 | VTE发病率[%(患病例数/总例数)] | OR值(95%CI) | P值 | |
|---|---|---|---|---|---|---|---|---|---|
| 伴基因突变者 | 不伴基因突变者 | ||||||||
| Tang[17],2012 | 中国 | PROC | c.565C>T(p.Arg189Trp) | rs146922325 | 蛋白C轻链C末端的21个连接肽 | 5.88(59/1 003) | 0.87(9/1 031) | 7.10(3.50~14.39) | <0.001 |
| Tang[32],2013 | 中国 | 同上 | 同上 | 同上 | 同上 | 5.21(68/1 304) | 0.90(12/1 334) | 6.06(3.26~11.25) | <0.001 |
| Tang[20],2012 | 中国 | PROC | c.574_576del(p.Lys193del) | rs199469469 | 蛋白C轻链C末端的21个连接肽 | 6.78(68/1 003) | 2.42(25/1 031) | 2.93(1.84~4.67) | <0.001 |
| Tang[32],2013 | 中国 | 同上 | 同上 | 同上 | 同上 | 6.52(85/1 304) | 2.40(32/1 334) | 2.84(1.88~4.29) | <0.001 |
| Kinoshita[15],2005 | 日本 | PROS1 | c.586A>G(p.Lys196Glu) | rs121918474 | 蛋白S第二表皮生长因子样结构域 | 5.88(5/85) | 1.64(5/304) | 3.74(1.06~13.20) | — |
| Ikejiri[23],2010 | 日本 | 同上 | 同上 | 同上 | 同上 | 10.00(6/60) | 1.28(3/234) | 8.56(2.07~35.30) | <0.05 |
| Kim[3],2013 | 韩国 | PROC | c.631C>T(p.Arg211Trp) | — | 蛋白C重链的α凝血酶裂解位点的CpG热点 | 2.80(14/500) | 0.24(7/2 953) | — | — |
| Kim[3],2013 | 韩国 | PROC | c.1218G>A(p.Met406Ile) | — | 蛋白C胰蛋白酶样丝氨酸蛋白酶结构域非CpG位点 | 1.80(9/500) | 0(0/2 953) | — | — |
注:"—"表示文献未提及相关数据;VTE为静脉血栓栓塞症,SNP为单核苷酸多态性
目前,遗传性蛋白C和蛋白S缺乏症的治疗方式主要有抗凝蛋白的替代治疗和抗凝治疗。替代疗法包括输注新鲜冷冻血浆(fresh frozen plasma,FFP)和外源性抗凝蛋白,如蛋白C浓缩物[34,35]。VTE患者以抗凝治疗为主,通常采用肝素或者其衍生物进行肠、胃外抗凝治疗。华法林可与低分子量肝素同时给药,直至连续2 d国际标准化比率(international normalized ratio,INR)达2.0~3.0,即可停用华法林,以防止华法林治疗期间出现罕见的皮肤坏死不良反应[36]。直接口服抗凝剂(direct oral anticoagulants,DOAC),如利伐沙班和阿哌沙班,可与FⅩa结合并阻止后者将凝血酶原裂解为凝血酶,接受该类药物治疗的患者复发和出血风险较低,是VTE患者的新治疗选择之一[37,38]。目前,DOAC在抗凝蛋白缺乏症患者中的疗效尚不明确,仍需进一步研究以确认DOAC长期疗效[39]。严重遗传性蛋白C缺乏症患者的治疗方法是肝移植,但是目前仅有少数采用肝移植成功治疗遗传性蛋白C缺乏症的病例报道[40]。此外,大多数蛋白C和蛋白S杂合突变的携带者无明显症状,临床主要采用观察治疗和预防首次血栓形成,不需要特殊治疗。这类患者合并其他遗传或者获得性危险因素,如FⅤ Leiden,抗磷脂抗体阳性或者外伤时需要进行VTE的预防性治疗。伴VTE复发高风险的患者需要维持长期的抗凝治疗。
基因治疗是指将外源基因导入靶细胞,以治疗突变或者缺陷基因引起的特定疾病。遗传性蛋白C和蛋白S缺乏症是由基因突变引起的疾病,大多数突变都是单碱基改变导致的错义或者无义突变。在结构上蛋白C和蛋白S类似于其他维生素K依赖性的凝血因子,包括凝血酶原、FⅦ、FⅨ和FⅩ。其中F9基因突变引起的B型血友病(hemophilia B,HB)是基因治疗的理想模型,因为FⅨ表达水平的小幅增加就能显著改善严重患者的出血表型,而且FⅨ治疗的有效范围较宽,不需对FⅨ表达水平进行严格控制。使用腺相关病毒(adeno-associated virus,AAV)载体表达人FⅨ的基因治疗已取得了巨大的进展[41,42]。在针对HB的肝定向基因疗法的研究中,10例HB患者(FⅨ活性低于正常参考值范围)进行AAV载体输注后持续表达FⅨ,FⅨ活性为(33.7±18.5)%[41]。在另一项基于AAV5载体介导F9基因转移的临床试验中,9例HB患者在接受单次给药后持久性表达FⅨ,自发性出血和FⅨ浓缩制剂的使用显著减少,并且未检测到针对AAV5衣壳的细胞免疫应答[42]。因此,基因治疗是治愈遗传性蛋白C和蛋白S缺乏症的理想策略,具有广阔的应用前景,值得进一步深入研究。
综上所述,遗传因素在VTE的发展中发挥至关重要的作用,亚洲人群VTE的主要遗传危险因素是抗凝蛋白的遗传异常,尤其是在APC抗凝系统中发挥重要作用蛋白C和蛋白S的异常。目前,相关研究已确定了遗传性蛋白C和蛋白S缺乏症常见的基因突变和基因多态性,针对性检测常见突变基因,有利于提高VTE的诊断和预防水平。但是这些遗传危险因素常见于东亚国家人群,其是否是亚洲其他地区人群VTE的危险因素仍值得探讨。此外,与VTE相关的其他遗传因素仍需继续研究,全面了解VTE的遗传学特征,有助于最大限度降低VTE形成的风险。
所有作者均声明不存在利益冲突

























